7. The reduced competence caused by the compensatory mutations in the
sxy-6 strain proves that the
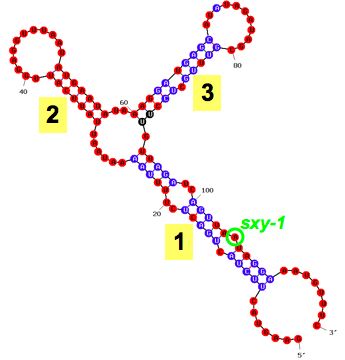
sxy-1 and
sxy-3 mutations cause hypercompetence by decreasing the number of base pairs in folded
sxy mRNA. To confirm the effect, and to see how strongly the folding can inhibit sxy expression, we made a mutant with two extra base pairs (sxy
-7). As predicted, it is even less competent - in fact it can't be transformed at all (Fig. 6A).
8. The
sxy-6 mutant isn't just not hypercompetent, it's much less competent than wildtype cells (10-fold to 500-fold, depending on the assay conditions), even though it is predicted to have the same number of base pairs when folded. This was especially surprising because its mutations replace a strong G:C basepair with a weaker A:T basepair. This may be telling us that in wildtype mRNA the G and C bases interact with bases at other positions as well as with each other, and that these unknown interactions enhance expression of
sxy.
9. We also directly checked the amounts of Sxy protein in the
sxy-6 and
sxy-7 mutants; it's much less than in the parent mutants and in wildtype cells (Fig. 6B). I don't have the draft version of Fig. 6B yet, although the data has been collected.
10. We don't know how the mRNA base pairing prevents
sxy expression, although we are considering several possibilities, all involving effects either on the amount of
sxy mRNA in the cell, or on the ability of the mRNA to be translated into protein. To distinguish between these, we examined the amounts of sxy mRNA in wildtype and mutant cells under various culture conditions. If folding reduces Sxy protein by reducing the amount of mRNA, then the mutant cells should have amounts of
sxy mRNA proportional to their amounts of Sxy protein. If folding acts only by preventing translation, then the mutants should all have the same amount of
sxy mRNA as wildtype cells.
The results are intermediate. The mutants contain less
sxy mRNA than the wildtype cells, but they contain more mRNA than we would expect from their reduction in Sxy protein (Fig. 7; I don't have all of the final data for this figure). This means that the folding must limit both the amount of
sxy mRNA and its ability to be translated into protein.
11. This conclusion is supported by data from gene fusions that put the
E. coli lacZ gene under control of
sxy mRNA. One kind of fusion ("operon fusion") tells us about effects of
sxy amount and on folding on mRNA amount; the other ("protein fusion") tells us about the combined effects on mRNAmRNA translatability. [
I hope I've got this right - I find it very hard to keep these effects straight.] We (
i.e. a former PhD student) constructed two pairs of fusions. In one pair, both fusions (protein and operon) join
lacZ gene (to a downstream position in the wildtype sxycodon 89), meaning that in both the mRNA can fold normally. In the other pair, both fusions join
lacZ to a position further upstream (codon 11) in the mRNA, bypassing a part of the mRNA essential for folding (so these fusion RNAs should have no folding).
Caveat: I don't know whether we've checked the predicted folding of the codon 11 fusion mRNAs. It will lack the base pairings involved in the mutations we've studied (the main stem), but could still have the other 'internal' base pairs that we think might block access to the translation start site.
After much tearing of hair and drawing on the whiteboard, I think it's clearest to consider the no-folding fusions first, and then examine how folding changes lacZ
expression (measured as beta-galactosidase activity). The no-folding protein fusion produces 2.8 times as much beta-galactosidase as the no-folding operon fusion. We expect both to have the same amount of mRNA, so this means that the
sxy translational signals (ribosome-binding site and start codon) work 2.8 times better than the
lacZones. That's neither surprising nor interesting in this context. The folding operon fusion produces only 0.45 as much activity as the corresponding no-folding fusion, telling us that folding reduces the amount of mRNA to 0.45. The folding protein fusion produces only 0.20 as much activity as the comparable no-folding fusion. We expect the folding fusion to have produced 0.45 as much mRNA, so the folded mRNA must be translated only 0.44 as well as the no-folding mRNA (Fig. 8).
It will be nice if the direct measurements of mRNA and Sxy protein (by real-time PCR and immunoblot respectively; Fig. 7) give the same magnitude of effects as the fusions. The preliminary data for Fig. 7 suggests a stronger effect of translatability than the fusions suggest.
13. Work by another group about 10 years ago had suggested that
sxy transcription is stimulated by cAMP, and they reported that the
sxy promoter contained a CRP binding site responsible for this effect. We find that adding cAMP
reduces expression of the sxy operon fusion. This is consistent with the location of a CRP site in a position where it will block transcription rather than stimulating it. I don't think this result deserves a figure (maybe because we don't have pretty data).
14. We did some microarray analysis of expression of all genes in the
sxy-1 hypercompetent mutant, under conditions that don't induce competence in wildtype cells. We can mention that all the competence regulon (CRP-S regulon) genes were being overexpressed. We won't show the data as we haven't done lots of microarray replicates (maybe we only did one). This should probably be described briefly under point 2 in the previous post.
15. We have examined the predicted foldings of
sxy mRNAs from related species in the family Pasteurellaceae. None of them were predicted to fold like
H. influenzae sxy. This could just be mentioned in the Discussion.