We're collaborating with some physicists on a cool laser-tweezers experiment. The tests are done on single cells, so it's important that the cells used for this experiment are as competent as possible. Our collaborator just asked " Is there some quantitative way to go between the transformation frequency of a culture and the fraction of cells that are competent, so we'll know, for a particular batch of cells, how many we need to try?
Yes, and it's a very simple old-fashioned experiment.
Cells that are maximally competent take up at least several DNA fragments. If we give them chromosomal DNA carrying two different antibiotic resistance genes (say novR and nalR), some cells will become resistant to both antibiotics because they took up both genes (double transformants). For this analysis it's important that the two genes be far enough apart on the chromosome that they will be in different DNA fragments. If all the cells are competent, we can predict the frequency of these double transformants; it's the product of the frequencies of single transformants. So if 1% (0.01 or 10^-2) of the cells in this experiment became NovR, and 0.5% (0.005 or 5x10^-3) of the cells became NalR, we predict that 0.005% (0.00005 or 5x10^-5) of the cells will be NovR NalR.
But what if not all the cells were competent? Say only 10% of them were. Then the 1% transformation frequency to NovR was because 10% of the cells had a transformation frequency of 10% and 90% had a transformation frequency of 0. Similarly the 0.5% transformation frequency to NalR would be because 10% of the cells had a transformation frequency of 5% and 90% of the cells had a transformation frequency of 0. Now the predicted frequency of double transformants is different. The 10% that are competent will have a double transformation frequency of 0.5%, and the 90% not competent a frequency of 0, giving a total frequency of 0.05%. This is ten-fold higher than if all the cells were competent.
So, a double transformant frequency higher than expected if all the cells were competent means that only a fraction of the cells were competent. On the other hand, if all cells were only a bit competent, so most cells were only taking up a single fragment, the same analysis would give a double transformation frequency lower than expected. This was all worked out 30 or more years ago.
When we do this analysis on competent H. influenzae cells, we always find that the double transformant frequency is higher than expected if all the cells were competent, which means that only some of the cells are competent, and that the competent cells are taking up multiple DNA fragments. (For reasons having to do with how the DNA is integrated into the chromosome, there's a fudge factor of 2.) When the transformation frequency for the single markers is high, the calculations indicate that about half of the cells are taking up DNA. When the single-transformant frequency is low (in some mutants, and in cells from conditions that only weakly induce competence), fewer cells are competent, but the ones that are are still taking up multiple DNA fragments.
So, we've usually used the single-marker transformation frequency as an indicator of the fraction of the cells that are competent. Maybe for the laser-tweezers experiments we should check again.
Our vindication is on hold...
The result I was raving about a couple of weeks ago turns out to not be as solid as I had thought. This was the comparison of hypercompetent cells with and without the purine-binding repressor PurR. The result appeared to show that getting rid of the repressor let the cells become much more competent, solidifying our hypothesis that cells turn on competence genes because they run out of purines.
There were two problems with the results. First, the PurR+ and PurR- cells were tested on different days, not in a single experiment. This means that uncontrollable variables could have been responsible for the difference. Second and worse, an important controllable variable was changed in the second experiment; the PurR- cells were treated with a lower concentration of purines than was used for the Pur+ cells.
The obvious correction is to repeat the experiment, with both cell types treated with the same concentration. This has now been done, and the exciting difference largely disappeared.
We still think that the purine repressor does repress one of the competence genes, as the gene's promoter has what appears to be a very good binding site for the repressor. But the analysis is confounded by the complexities of purine interconversion in the cytoplasm, and by the toxicity of high purine concentrations for the cells. This means we probably won't get a clean answer until we've done a lot more experiments.
There were two problems with the results. First, the PurR+ and PurR- cells were tested on different days, not in a single experiment. This means that uncontrollable variables could have been responsible for the difference. Second and worse, an important controllable variable was changed in the second experiment; the PurR- cells were treated with a lower concentration of purines than was used for the Pur+ cells.
The obvious correction is to repeat the experiment, with both cell types treated with the same concentration. This has now been done, and the exciting difference largely disappeared.
We still think that the purine repressor does repress one of the competence genes, as the gene's promoter has what appears to be a very good binding site for the repressor. But the analysis is confounded by the complexities of purine interconversion in the cytoplasm, and by the toxicity of high purine concentrations for the cells. This means we probably won't get a clean answer until we've done a lot more experiments.
Ramifying motif searches
I began using the Gibbs motif search program just to get a better estimate of 'the' USS consensus. But as I get into it (necessary to get it working effectively) I'm discovering more and more questions I can answer with it. Here are what I have so far:
1. Does the direction in which a sequence is replicated affect the consensus of its USSs? That is, do USSs whose "forward" orientation sequence is found in the leading strand during DNA replication have a slightly different consensus than those whose "reverse" orientation sequence is in the leading strand? I raised this issue at the end of an earlier post, but I haven't tested it yet. All I need to do is to get the sequence of each strand of the genome, chop them ar the origin and terminus of replication, and then put the parts together so all the leading strand sequences are in one file, and all the lagging strand sequences are in another file. Then run both files through the motif search.
2. For USSs in coding regions (this is most USSs), does their orientation with respect to the direction of transcription and translation affect their consensus? This seemed very easy to do, as I was able to download from TIGR a file containing the nucleotide sequences of all the H. influenzae coding regions. I had to tidy away the small number of non-ACGT bases (I replaced them with Ns, using Word) and thought I was good to go. I would just have the program search for the forward motif and for the reverse motif, and see if the two motifs were complementary. But for some reason the motif-search program has a much harder time finding a motif in the set of gene sequences than in the set of whole-genome sequences. I'm beginning to suspect that it prefers its sequences in big fragments rather than in gene-sized pieces. This seems a bit improbable, so I'll wait until I've tested it before emailing the expert for help. Tomorrow I'll test it by getting the post-doc to use her clever Perl program to chop the genome into 500bp fragments (in Fasta format), and compare how the program handles this to the usual 9kb fragments.
 3. Do the bases at different positions in the motif interact with each other in a way that makes some combinations more likely than others? Put another way, in the aligned set of sequences the motif search produces, position 16 is a T 20% of the time, and position 17 is a T 44% of the time. If all else is equal, we'd expect both positions to be Ts 8.8% of the time. But if the effects of each T work together well, in a way that, for example, an A and a T don't, then more than 8.8% of the sequences might have TTs here. We can use programs designed to detect the kind of genetic interactions called 'linkage disequilibrium' to detect these effects.
3. Do the bases at different positions in the motif interact with each other in a way that makes some combinations more likely than others? Put another way, in the aligned set of sequences the motif search produces, position 16 is a T 20% of the time, and position 17 is a T 44% of the time. If all else is equal, we'd expect both positions to be Ts 8.8% of the time. But if the effects of each T work together well, in a way that, for example, an A and a T don't, then more than 8.8% of the sequences might have TTs here. We can use programs designed to detect the kind of genetic interactions called 'linkage disequilibrium' to detect these effects.
We didn't know what program(s) to use, so we posted a brief description of what we wanted to do onto the 'Evoldir' emailing list, where it would be seen by almost every evolutionary biologist on the planet. We got four very helpful replies, and soon I'll understand the programs well enough to answer our question.
1. Does the direction in which a sequence is replicated affect the consensus of its USSs? That is, do USSs whose "forward" orientation sequence is found in the leading strand during DNA replication have a slightly different consensus than those whose "reverse" orientation sequence is in the leading strand? I raised this issue at the end of an earlier post, but I haven't tested it yet. All I need to do is to get the sequence of each strand of the genome, chop them ar the origin and terminus of replication, and then put the parts together so all the leading strand sequences are in one file, and all the lagging strand sequences are in another file. Then run both files through the motif search.
2. For USSs in coding regions (this is most USSs), does their orientation with respect to the direction of transcription and translation affect their consensus? This seemed very easy to do, as I was able to download from TIGR a file containing the nucleotide sequences of all the H. influenzae coding regions. I had to tidy away the small number of non-ACGT bases (I replaced them with Ns, using Word) and thought I was good to go. I would just have the program search for the forward motif and for the reverse motif, and see if the two motifs were complementary. But for some reason the motif-search program has a much harder time finding a motif in the set of gene sequences than in the set of whole-genome sequences. I'm beginning to suspect that it prefers its sequences in big fragments rather than in gene-sized pieces. This seems a bit improbable, so I'll wait until I've tested it before emailing the expert for help. Tomorrow I'll test it by getting the post-doc to use her clever Perl program to chop the genome into 500bp fragments (in Fasta format), and compare how the program handles this to the usual 9kb fragments.
 3. Do the bases at different positions in the motif interact with each other in a way that makes some combinations more likely than others? Put another way, in the aligned set of sequences the motif search produces, position 16 is a T 20% of the time, and position 17 is a T 44% of the time. If all else is equal, we'd expect both positions to be Ts 8.8% of the time. But if the effects of each T work together well, in a way that, for example, an A and a T don't, then more than 8.8% of the sequences might have TTs here. We can use programs designed to detect the kind of genetic interactions called 'linkage disequilibrium' to detect these effects.
3. Do the bases at different positions in the motif interact with each other in a way that makes some combinations more likely than others? Put another way, in the aligned set of sequences the motif search produces, position 16 is a T 20% of the time, and position 17 is a T 44% of the time. If all else is equal, we'd expect both positions to be Ts 8.8% of the time. But if the effects of each T work together well, in a way that, for example, an A and a T don't, then more than 8.8% of the sequences might have TTs here. We can use programs designed to detect the kind of genetic interactions called 'linkage disequilibrium' to detect these effects.We didn't know what program(s) to use, so we posted a brief description of what we wanted to do onto the 'Evoldir' emailing list, where it would be seen by almost every evolutionary biologist on the planet. We got four very helpful replies, and soon I'll understand the programs well enough to answer our question.
Make the bacteria do the work
The best geneticists are smart but lazy; they use genetics to avoid laborious and tedious analyses, and in the process produce very elegant experiments, generating the maximum information with the minimum work. Our biochemical analysis of how sxy mRNA folds has been a lot of work but I don't feel that we've learned much more than we already knew (from genetics), and I'm thinking it's time to give genetics another chance.
We discovered (and named) sxy as the site of a mutation that causes cells to be competent when normal cells aren't. We now have five such mutations, but when we started we didn't even know if such mutations could arise. But it was relatively easy to find out, using a genetic selection experiment.
We took normal cells, gave them a small dose of a mutagen to increase the number of cells containing new mutations, and grew them under conditions where normal cells wouldn't become competent. Then we gave them some DNA with an antibiotic-resistance gene, and put them on agar plates containing that antibiotic. Normal cells die on these plates because, not being competent, they can't take up the antibiotic-resistance gene. So cells that do survive this selection are likely to have a mutation that changes how competence is regulated.
Note that we didn't have to test all the cells we started with, to see if they might have the kind of mutation we were looking for. We just gave them DNA, put them on the antibiotic plates, and let them find out for themselves whether they were the lucky one in a billion that had a mutation that kept them alive. That's what I mean by letting the bacteria do the work.
We've had these five 'hypercompetence' mutations for a long time (more than 10 years!). Because four of them wouldn't change the Sxy protein, but each of them would prevent a base pair from forming when two separate parts of the sxy mRNA came together, we hypothesized that RNA base pairing limits expression of the sxy gene.
A former grad student did the definitive test by making 'compensatory mutations'. These are mutations that restore the base pairing without restoring the wildtype DNA sequence. The most elegant one she made combined two independent hypercompetence mutations that affected the two partners in the same hypothesized base pair. The original mutations each prevented the base pair from forming, but the double mutant allowed the mutant bases to pair with each other. If base pairing wasn't important the double mutant should be even MORE hypercompetent that the single mutants, but if the single mutants caused hypercompetence by preventing base pairing, the double mutant should be much LESS hypercompetent, and that's what she found.
We now know more about the consequences of these mutations. They affect both the amount of sxy mRNA and the amount of Sxy protein made from each sxy mRNA, and they reduce the ability of purine nucleotide pools to limit sxy expression. But we don't know anything more about how the mutations do this, despite a lot of work trying to directly examine the folding of sxy mRNA.
So I think it's time to go back to genetics, and look for more mutations in sxy. We still have some of the original mutagenized cells in the freezer, and we could repeat the original selection for hypercompetent mutants on these, or on a fresh batch of mutagenized cells. We could also limit our mutagenesis to the sxy gene, by randomly mutagenizing a cloned or PCR-amplified sxy gene and then transforming it into normal cells and selecting for hypercompetence mutants. The results of such a mutant hunt could identify other parts of the mRNA that participate in regulatory base pairing, or that otherwise limit transcription or translation (perhaps by interacting with a purine).
Unfortunately selection for hypercompetent mutants won't give us the other kind of regulatory mutations, those that reduce sxy expression. In principle we could set up the reverse selection, a treatment that would kill all the cells that did take up DNA, but this would require having some sort of 'killer gene'. Such genes exist in other systems, but using the same principles to select for cells that can't become competent is likely to be very difficult or impossible.
But given that we are only interested in mutations in sxy (for this specific problem, not all our research), we can invest the time to do a genetic screen instead of a selection. That is, we'll have to do more of the work ourselves. We don't want to make random mutations throughout the genome, as many genes other than sxy are needed for DNA uptake. Techniques are available (kits!) to create specific mutations at any desired positions, and we can use these to create a set of many different mutations in sxy. We can then screen each mutant separately for the ability to take up DNA. Luckily the sxy gene is small, and we can use the results of our selection for hypercompetent mutants to guide this screen.
We discovered (and named) sxy as the site of a mutation that causes cells to be competent when normal cells aren't. We now have five such mutations, but when we started we didn't even know if such mutations could arise. But it was relatively easy to find out, using a genetic selection experiment.
We took normal cells, gave them a small dose of a mutagen to increase the number of cells containing new mutations, and grew them under conditions where normal cells wouldn't become competent. Then we gave them some DNA with an antibiotic-resistance gene, and put them on agar plates containing that antibiotic. Normal cells die on these plates because, not being competent, they can't take up the antibiotic-resistance gene. So cells that do survive this selection are likely to have a mutation that changes how competence is regulated.
Note that we didn't have to test all the cells we started with, to see if they might have the kind of mutation we were looking for. We just gave them DNA, put them on the antibiotic plates, and let them find out for themselves whether they were the lucky one in a billion that had a mutation that kept them alive. That's what I mean by letting the bacteria do the work.
We've had these five 'hypercompetence' mutations for a long time (more than 10 years!). Because four of them wouldn't change the Sxy protein, but each of them would prevent a base pair from forming when two separate parts of the sxy mRNA came together, we hypothesized that RNA base pairing limits expression of the sxy gene.
A former grad student did the definitive test by making 'compensatory mutations'. These are mutations that restore the base pairing without restoring the wildtype DNA sequence. The most elegant one she made combined two independent hypercompetence mutations that affected the two partners in the same hypothesized base pair. The original mutations each prevented the base pair from forming, but the double mutant allowed the mutant bases to pair with each other. If base pairing wasn't important the double mutant should be even MORE hypercompetent that the single mutants, but if the single mutants caused hypercompetence by preventing base pairing, the double mutant should be much LESS hypercompetent, and that's what she found.
We now know more about the consequences of these mutations. They affect both the amount of sxy mRNA and the amount of Sxy protein made from each sxy mRNA, and they reduce the ability of purine nucleotide pools to limit sxy expression. But we don't know anything more about how the mutations do this, despite a lot of work trying to directly examine the folding of sxy mRNA.
So I think it's time to go back to genetics, and look for more mutations in sxy. We still have some of the original mutagenized cells in the freezer, and we could repeat the original selection for hypercompetent mutants on these, or on a fresh batch of mutagenized cells. We could also limit our mutagenesis to the sxy gene, by randomly mutagenizing a cloned or PCR-amplified sxy gene and then transforming it into normal cells and selecting for hypercompetence mutants. The results of such a mutant hunt could identify other parts of the mRNA that participate in regulatory base pairing, or that otherwise limit transcription or translation (perhaps by interacting with a purine).
Unfortunately selection for hypercompetent mutants won't give us the other kind of regulatory mutations, those that reduce sxy expression. In principle we could set up the reverse selection, a treatment that would kill all the cells that did take up DNA, but this would require having some sort of 'killer gene'. Such genes exist in other systems, but using the same principles to select for cells that can't become competent is likely to be very difficult or impossible.
But given that we are only interested in mutations in sxy (for this specific problem, not all our research), we can invest the time to do a genetic screen instead of a selection. That is, we'll have to do more of the work ourselves. We don't want to make random mutations throughout the genome, as many genes other than sxy are needed for DNA uptake. Techniques are available (kits!) to create specific mutations at any desired positions, and we can use these to create a set of many different mutations in sxy. We can then screen each mutant separately for the ability to take up DNA. Luckily the sxy gene is small, and we can use the results of our selection for hypercompetent mutants to guide this screen.
Folding variations
The server I've been using for USS motif searches has been down, so I've been playing around with variations in how sxy mRNA might fold. By understanding the variations that occur naturally we hope to get insight into the mechanism by which the folding regulates sxy expression, and also how natural selection has been acting on this mechanism.
The folding program Mfold presents only one structure (the first one below) when it folds the sxy mRNA regulatory region (the 50 non-coding bases upstream of the start codon and the first 60 bases of the coding region). I guess this is because other possible foldings are predicted to be much less stable. To get insight into alternative foldings I tried changing the input sequence by making it shorter or longer at the 5' or 3' end. Changing the 5' end might reflect consequences of using different transcription start sites or of binding of a ribosome to its site in front of the start codon. Changing the 3' end might mimic the effect of RNA polymerase stalling or progressing quickly along the DNA. The length changes might also just reveal whether interactions between particular parts of the RNA were sensitive to the presence of other potentially interacting parts.
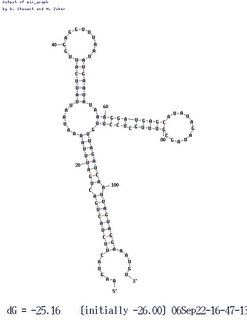 Result: There are indeed no alternative stable foldings for RNAs that differ only in small ways from the known regulatory region. However if the RNA is shifted more substantially to include only coding sequences (i.e. the non-coding part of the RNA is not included, and the coding part is extended), another stable folding emerges. This could be biologically significant, if its ability to compete with the 'standard' folding depends on either ribosome binding or rate or transcription. But the factors involved are so complex that I'm starting to see more 'mutant hunts' as our best approach. I'll write about them in a separate post.
Result: There are indeed no alternative stable foldings for RNAs that differ only in small ways from the known regulatory region. However if the RNA is shifted more substantially to include only coding sequences (i.e. the non-coding part of the RNA is not included, and the coding part is extended), another stable folding emerges. This could be biologically significant, if its ability to compete with the 'standard' folding depends on either ribosome binding or rate or transcription. But the factors involved are so complex that I'm starting to see more 'mutant hunts' as our best approach. I'll write about them in a separate post.
Thanks to the genome-sequencing efforts of colleagues, we also have sxy sequences from four other (independent) isolates of H. influenzae. Three of these are identical to our KW20 strain, so of course the folding software treats them identically, but the fourth differs at about half a dozen positions. Three of these differences are in the regulatory region, and they cause an interesting change in folding (shown here). Two of the differences eliminate base pairs in the 'standard' folding, and a third is in an unpaired loop. The new folding puts two of these changed bases into new base pairs, and this folded RNA is predicted to be slightly MORE thermodynamically stable than the standard folding (deltaG = 25.5 rather than 23.1).
dozen positions. Three of these differences are in the regulatory region, and they cause an interesting change in folding (shown here). Two of the differences eliminate base pairs in the 'standard' folding, and a third is in an unpaired loop. The new folding puts two of these changed bases into new base pairs, and this folded RNA is predicted to be slightly MORE thermodynamically stable than the standard folding (deltaG = 25.5 rather than 23.1).
In the variant folding the critical stem identified by our hypercompetence mutations is unchanged, but the big loop of RNA that includes the ribosome-binding site and the start codon is folded very differently. However the folding still blocks access of the ribosome to its site, although in a different way. This may be telling us that blocking the ribosome is an important function of the folding. That is, it may be part of the mechanism by which folding of the mRNA regulates its stability or its ability to be translated into protein.
The folding program Mfold presents only one structure (the first one below) when it folds the sxy mRNA regulatory region (the 50 non-coding bases upstream of the start codon and the first 60 bases of the coding region). I guess this is because other possible foldings are predicted to be much less stable. To get insight into alternative foldings I tried changing the input sequence by making it shorter or longer at the 5' or 3' end. Changing the 5' end might reflect consequences of using different transcription start sites or of binding of a ribosome to its site in front of the start codon. Changing the 3' end might mimic the effect of RNA polymerase stalling or progressing quickly along the DNA. The length changes might also just reveal whether interactions between particular parts of the RNA were sensitive to the presence of other potentially interacting parts.
 Result: There are indeed no alternative stable foldings for RNAs that differ only in small ways from the known regulatory region. However if the RNA is shifted more substantially to include only coding sequences (i.e. the non-coding part of the RNA is not included, and the coding part is extended), another stable folding emerges. This could be biologically significant, if its ability to compete with the 'standard' folding depends on either ribosome binding or rate or transcription. But the factors involved are so complex that I'm starting to see more 'mutant hunts' as our best approach. I'll write about them in a separate post.
Result: There are indeed no alternative stable foldings for RNAs that differ only in small ways from the known regulatory region. However if the RNA is shifted more substantially to include only coding sequences (i.e. the non-coding part of the RNA is not included, and the coding part is extended), another stable folding emerges. This could be biologically significant, if its ability to compete with the 'standard' folding depends on either ribosome binding or rate or transcription. But the factors involved are so complex that I'm starting to see more 'mutant hunts' as our best approach. I'll write about them in a separate post.Thanks to the genome-sequencing efforts of colleagues, we also have sxy sequences from four other (independent) isolates of H. influenzae. Three of these are identical to our KW20 strain, so of course the folding software treats them identically, but the fourth differs at about half a
 dozen positions. Three of these differences are in the regulatory region, and they cause an interesting change in folding (shown here). Two of the differences eliminate base pairs in the 'standard' folding, and a third is in an unpaired loop. The new folding puts two of these changed bases into new base pairs, and this folded RNA is predicted to be slightly MORE thermodynamically stable than the standard folding (deltaG = 25.5 rather than 23.1).
dozen positions. Three of these differences are in the regulatory region, and they cause an interesting change in folding (shown here). Two of the differences eliminate base pairs in the 'standard' folding, and a third is in an unpaired loop. The new folding puts two of these changed bases into new base pairs, and this folded RNA is predicted to be slightly MORE thermodynamically stable than the standard folding (deltaG = 25.5 rather than 23.1).In the variant folding the critical stem identified by our hypercompetence mutations is unchanged, but the big loop of RNA that includes the ribosome-binding site and the start codon is folded very differently. However the folding still blocks access of the ribosome to its site, although in a different way. This may be telling us that blocking the ribosome is an important function of the folding. That is, it may be part of the mechanism by which folding of the mRNA regulates its stability or its ability to be translated into protein.
Deconstructing the structure?
A lab meeting presentation yesterday got me worrying about how having an explicit technical drawing of a structure makes it difficult to think about alternative structures.
We started 10 or 12 years ago with a hand-drawn sketch of how parts of sxy mRNA might pair with each other. But now we've been relying on a computer program called Mfold, which takes any RNA sequence and uses thermodynamic principles to deduce the most stable configurations it could fold into. It gives nice professional-looking drawings, and I'm afraid we're being seduced by them into not fully considering the other possibilities.
It's not Mfold's fault. Mfold offers several alternative structures, but it's all too easy to focus on the one with the highest thermodynamic stability score and ignore the others. This would be the right thing to do if we knew that the score correctly predicted the real structure of the RNA in the cell, but there are lots of reasons to doubt this.
First, the thermodynamics is only an approximation of reality. Second, the foldings are too simple - each position interacts with at most one other position - whereas far more complex interactions are likely to occur in real molecules. Third, interactions are dynamic and stochastic; a population of RNA molecules will include many different conformations, and each molecule's conformation is likely to fluctuate over time. Fourth, many biologically significant effects of secondary structures depend on competition between two or more different possible foldings, as in the following made-up example. Part B of an RNA strand can either pair with part A or with part C. If it pairs with A, the ribosome-binding site in part C is available and the protein can be made, but if it pairs with C the site is blocked and no protein is made.
We have genetic evidence for one short section of pairing in sxy mRNA, between sequences separated by about 90 bases. This is very solid; we know which bases pair with which in this section. But what about the rest of the molecule, especially the intervening 90 bases? We now have biochemical (RNase digestion) evidence over the whole region. This tells us that some parts of the RNA are in a base-paired state, but it doesn't tell us which other bases they are paired with. It also tells us that some bases are not base-paired, and it gives us no information about the states of still other bases, The 'paired' and 'not paired' states match quite well to those predicted by the most stable Mfold structure, but I think we should spend a bit more time considering whether they also fit alternative foldings. Alternatives include other Mfold predictions, predictions from other programs, or even structures we've come up with ourselves, folding 'by eye' rather than by computer.
We started 10 or 12 years ago with a hand-drawn sketch of how parts of sxy mRNA might pair with each other. But now we've been relying on a computer program called Mfold, which takes any RNA sequence and uses thermodynamic principles to deduce the most stable configurations it could fold into. It gives nice professional-looking drawings, and I'm afraid we're being seduced by them into not fully considering the other possibilities.
It's not Mfold's fault. Mfold offers several alternative structures, but it's all too easy to focus on the one with the highest thermodynamic stability score and ignore the others. This would be the right thing to do if we knew that the score correctly predicted the real structure of the RNA in the cell, but there are lots of reasons to doubt this.
First, the thermodynamics is only an approximation of reality. Second, the foldings are too simple - each position interacts with at most one other position - whereas far more complex interactions are likely to occur in real molecules. Third, interactions are dynamic and stochastic; a population of RNA molecules will include many different conformations, and each molecule's conformation is likely to fluctuate over time. Fourth, many biologically significant effects of secondary structures depend on competition between two or more different possible foldings, as in the following made-up example. Part B of an RNA strand can either pair with part A or with part C. If it pairs with A, the ribosome-binding site in part C is available and the protein can be made, but if it pairs with C the site is blocked and no protein is made.
We have genetic evidence for one short section of pairing in sxy mRNA, between sequences separated by about 90 bases. This is very solid; we know which bases pair with which in this section. But what about the rest of the molecule, especially the intervening 90 bases? We now have biochemical (RNase digestion) evidence over the whole region. This tells us that some parts of the RNA are in a base-paired state, but it doesn't tell us which other bases they are paired with. It also tells us that some bases are not base-paired, and it gives us no information about the states of still other bases, The 'paired' and 'not paired' states match quite well to those predicted by the most stable Mfold structure, but I think we should spend a bit more time considering whether they also fit alternative foldings. Alternatives include other Mfold predictions, predictions from other programs, or even structures we've come up with ourselves, folding 'by eye' rather than by computer.
The first good searches for USS motifs
With more help from the motif-software expert, I now have the program running well. I can use fragmentation masks to control where the program puts gaps, and this also lets me set whether the search is for motifs in the 'forward' or 'reverse' orientation. I can search for motifs with up to 21 significant positions, spread over any length of sequence I care to specify.
I've run a couple of passes through the entire genome. The results are just as unsurprising as I had expected (is that a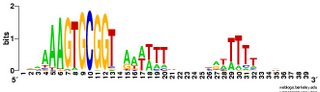 tautology?). Blogger won't upload the logo image files right now so I'll add them later (done). Basically, the forward and reverse motifs are fairly close reverse complements of each other. I was going to write that this means that there are no strong effects of the direction of DNA replication, but that's not true. To see effects of the direction of
tautology?). Blogger won't upload the logo image files right now so I'll add them later (done). Basically, the forward and reverse motifs are fairly close reverse complements of each other. I was going to write that this means that there are no strong effects of the direction of DNA replication, but that's not true. To see effects of the direction of  DNA replication I'd need to split the genome sequence into two parts, one clockwise from the origin of replication to the probable terminus, and one counterclockwise from the origin to the terminus and compare the forward and reverse motifs in each half. That way the forward/clockwise and referse/counterclockwise motifs would be derived from the 'leading' strand and the reverse/clockwise and forward/counterclockwise motifs would be derived from the lagging strand. [Or vice versa, as I don't know which strand is which.] Hmm, I don't think anyone has ever done this. So I should.
DNA replication I'd need to split the genome sequence into two parts, one clockwise from the origin of replication to the probable terminus, and one counterclockwise from the origin to the terminus and compare the forward and reverse motifs in each half. That way the forward/clockwise and referse/counterclockwise motifs would be derived from the 'leading' strand and the reverse/clockwise and forward/counterclockwise motifs would be derived from the lagging strand. [Or vice versa, as I don't know which strand is which.] Hmm, I don't think anyone has ever done this. So I should.
I've run a couple of passes through the entire genome. The results are just as unsurprising as I had expected (is that a
 tautology?). Blogger won't upload the logo image files right now so I'll add them later (done). Basically, the forward and reverse motifs are fairly close reverse complements of each other. I was going to write that this means that there are no strong effects of the direction of DNA replication, but that's not true. To see effects of the direction of
tautology?). Blogger won't upload the logo image files right now so I'll add them later (done). Basically, the forward and reverse motifs are fairly close reverse complements of each other. I was going to write that this means that there are no strong effects of the direction of DNA replication, but that's not true. To see effects of the direction of 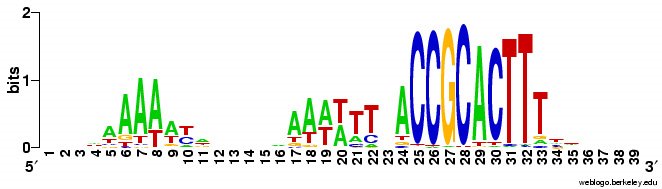 DNA replication I'd need to split the genome sequence into two parts, one clockwise from the origin of replication to the probable terminus, and one counterclockwise from the origin to the terminus and compare the forward and reverse motifs in each half. That way the forward/clockwise and referse/counterclockwise motifs would be derived from the 'leading' strand and the reverse/clockwise and forward/counterclockwise motifs would be derived from the lagging strand. [Or vice versa, as I don't know which strand is which.] Hmm, I don't think anyone has ever done this. So I should.
DNA replication I'd need to split the genome sequence into two parts, one clockwise from the origin of replication to the probable terminus, and one counterclockwise from the origin to the terminus and compare the forward and reverse motifs in each half. That way the forward/clockwise and referse/counterclockwise motifs would be derived from the 'leading' strand and the reverse/clockwise and forward/counterclockwise motifs would be derived from the lagging strand. [Or vice versa, as I don't know which strand is which.] Hmm, I don't think anyone has ever done this. So I should.
No-fault motif searching
Yesterday the very helpful guy who wrote the motif-search program I'm using sent me an improved version that eliminated the segmentation fault I'd been experiencing. Now the program will analyze a file containing the whole H. influenzae genome. He also sent advice on how to improve its accuracy by having it do many more trials, but I'm going to let this wait until we've solved the remaining problem.
That problem is the program's reluctance to fragment the motifs it finds (illustrated by the two logo images in the segmentation fault post). Because the program is optimized for finding the relatively compact motifs typical of sites where regulatory proteins bind, it prefers central positions with weak consensus over distant positions with strong consensuses.
The expert sent me instructions for specifying a "fragmentation mask" to overcome this. The mask is a string of numbers that specify the pattern of significant and nonsignificant positions in the desired motif. For example, 1110000111 specifies a motif with three significant positions on either side of four nonsignificant positions.
If I understand them correctly, masks can be used two ways, as starting suggestions or as strict rules. A mask with only zeros and ones is used as a suggestion, telling the program "Start with a motif that matches this pattern, but if you find a better pattern you can ignore the mask." A mask containing "3"s is used as a rule, with each 3 specifying a position that must remain nonsignificant in the final motif. For example, 1113333111 specifies a motif that must have four nonsignificant positions.
I'm trying to use a mask as a rule (with "3"s). But I suspect that I don't understand masks correctly, because the program reads my mask file but ignores the instructions in it. I have enough programming experience to know that this almost certainly means that my instructions are set up wrong. I've tried making the rule very simple (rather than the kind of rule I would use to force a USS-like fragmentation pattern), but even that doesn't work.
So I've emailed the helpful expert asking for more advice.
That problem is the program's reluctance to fragment the motifs it finds (illustrated by the two logo images in the segmentation fault post). Because the program is optimized for finding the relatively compact motifs typical of sites where regulatory proteins bind, it prefers central positions with weak consensus over distant positions with strong consensuses.
The expert sent me instructions for specifying a "fragmentation mask" to overcome this. The mask is a string of numbers that specify the pattern of significant and nonsignificant positions in the desired motif. For example, 1110000111 specifies a motif with three significant positions on either side of four nonsignificant positions.
If I understand them correctly, masks can be used two ways, as starting suggestions or as strict rules. A mask with only zeros and ones is used as a suggestion, telling the program "Start with a motif that matches this pattern, but if you find a better pattern you can ignore the mask." A mask containing "3"s is used as a rule, with each 3 specifying a position that must remain nonsignificant in the final motif. For example, 1113333111 specifies a motif that must have four nonsignificant positions.
I'm trying to use a mask as a rule (with "3"s). But I suspect that I don't understand masks correctly, because the program reads my mask file but ignores the instructions in it. I have enough programming experience to know that this almost certainly means that my instructions are set up wrong. I've tried making the rule very simple (rather than the kind of rule I would use to force a USS-like fragmentation pattern), but even that doesn't work.
So I've emailed the helpful expert asking for more advice.
Vindication!
About 15 years ago I hypothesized that Haemophilus influenzae takes up DNA mainly for its content of nucleotides, and predicted that the genes encoding the uptake machinery would be turned on when the cells' energy and nucleotide resources were depleted. At the time we had experimental evidence implicating an energy-depletion signal (CRP and cyclic AMP), but nothing implicating nucleotide pools.
About five years ago we reported that addition of purine nucleotides inhibited both transformation and the induction of two competence genes (comA and rec-2). This was consistent with my prediction, but because we didn't know anything about the molecular mechanism it wasn't compelling. As of last night we know a lot more about the mechanism.
Two different mechanisms are involved. First, nucleotides inhibit production of the competence activating protein Sxy, both by reducing both the amount of sxy mRNA and its ability to be translated into protein. This inhibition depends on base pairing in sxy mRNA; mutations that weaken the base pairing weaken the inhibition. Amp causes a stronger inhibition than GMP does. Second, when purines are abundant the purine repressor PurR represses one or more competence genes. Repression by PurR is stronger with GMP than with AMP, consistent with what's known about PurR's action in E. coli.
Of course this raises lots more questions (see below for my 'real number line' analogy).
What changes inside the cell when nucleotides are added to the outside? As I understand it, cells can't directly take up nucleotides because the phosphate has too strong a charge to cross the membrane, so why do nucleotides have a stronger effect than nucleosides? What molecule interacts with the sxymRNA base pairing? Does this reduce the amount of mRNA being made, or increase its breakdown? Does the molecule act at a riboswitch? Are micro-RNAs involved? Is coupling of transcription and translation involved? Does the same interaction inhibit translation of sxy mRNA, or is that a different effect? Does PurR repress transcription of the rec-2 gene? Does it repress any other competence genes? Why does adding cAMP partially counteract inhibition by GMP and, to a lesser extent, by GMP. Do the same regulatory effects occur in H. influenzae's relatives?
The real number line analogy: Scientific knowledge is like the "real number line" used in introductory math classes. In the line, every point is a number, but no matter how close together two points (or numbers) are, there are always infinitely many other points separating them. The real world similarly contains infinitely many things to discover. No matter how much we find out about something, there are always many more important things to find out. I use this analogy for beginning science students, who are often concerned that all the important discoveries have already been made.
About five years ago we reported that addition of purine nucleotides inhibited both transformation and the induction of two competence genes (comA and rec-2). This was consistent with my prediction, but because we didn't know anything about the molecular mechanism it wasn't compelling. As of last night we know a lot more about the mechanism.
Two different mechanisms are involved. First, nucleotides inhibit production of the competence activating protein Sxy, both by reducing both the amount of sxy mRNA and its ability to be translated into protein. This inhibition depends on base pairing in sxy mRNA; mutations that weaken the base pairing weaken the inhibition. Amp causes a stronger inhibition than GMP does. Second, when purines are abundant the purine repressor PurR represses one or more competence genes. Repression by PurR is stronger with GMP than with AMP, consistent with what's known about PurR's action in E. coli.
Of course this raises lots more questions (see below for my 'real number line' analogy).
What changes inside the cell when nucleotides are added to the outside? As I understand it, cells can't directly take up nucleotides because the phosphate has too strong a charge to cross the membrane, so why do nucleotides have a stronger effect than nucleosides? What molecule interacts with the sxymRNA base pairing? Does this reduce the amount of mRNA being made, or increase its breakdown? Does the molecule act at a riboswitch? Are micro-RNAs involved? Is coupling of transcription and translation involved? Does the same interaction inhibit translation of sxy mRNA, or is that a different effect? Does PurR repress transcription of the rec-2 gene? Does it repress any other competence genes? Why does adding cAMP partially counteract inhibition by GMP and, to a lesser extent, by GMP. Do the same regulatory effects occur in H. influenzae's relatives?
The real number line analogy: Scientific knowledge is like the "real number line" used in introductory math classes. In the line, every point is a number, but no matter how close together two points (or numbers) are, there are always infinitely many other points separating them. The real world similarly contains infinitely many things to discover. No matter how much we find out about something, there are always many more important things to find out. I use this analogy for beginning science students, who are often concerned that all the important discoveries have already been made.
Waiting for illumination...
The five double mutant strains I constructed appear to be correct. They should have the purR knockout combined with each of our five sxy hypercompetence mutations. I know they have the knockout because they're resistant to kanamycin (an insertion of the kanR gene is what knocks out purR). And I know they still have the sxy mutations, because I tested their competence under conditions that only slightly induce competence: the sxy+ purR- control had a transformation frequency of only 2 x 10^-7, but the sxy mutant purR- strains had transformation frequencies 100-500 times higher.
Now I'm waiting for the grad student's results of testing whether these mutants are now insensitive to the transformation-inhibiting effects of nucleotides. He finished the experiments last night, but won't know the results until the cells have had enough time to grow into colonies.
I'm hopeful, because my results already suggest our hypothesis is correct. As part of constructing the double mutants, I had measured the transformation frequencies of wildtype and sxy mutant cells (all purR+) under similar slightly-inducing conditions. The culture medium wasn't deliberately supplemented with nucleotides, but it's a 'rich' medium and we think that the nucleotides it naturally contains are part of the reason it allows only weak induction of competence. In this medium the sxy (purR+) mutants were only 4-10-fold more competent than the control sxy+ cells. Adding the purR knockout has made them 100-500 times more competent, suggesting that PurR does normally repress competence in this rich medium.
Now I'm waiting for the grad student's results of testing whether these mutants are now insensitive to the transformation-inhibiting effects of nucleotides. He finished the experiments last night, but won't know the results until the cells have had enough time to grow into colonies.
I'm hopeful, because my results already suggest our hypothesis is correct. As part of constructing the double mutants, I had measured the transformation frequencies of wildtype and sxy mutant cells (all purR+) under similar slightly-inducing conditions. The culture medium wasn't deliberately supplemented with nucleotides, but it's a 'rich' medium and we think that the nucleotides it naturally contains are part of the reason it allows only weak induction of competence. In this medium the sxy (purR+) mutants were only 4-10-fold more competent than the control sxy+ cells. Adding the purR knockout has made them 100-500 times more competent, suggesting that PurR does normally repress competence in this rich medium.
Part two of the puzzle

Iakov's comment to yesterday's post is right; the card images I posted yesterday fit a binary code. The card #9 we created worked fine in the scintillation counter.
But the simple binary code seems to fall apart for numbers higher than 10. Admittedly, we haven't worked very hard at solving it.
Not a genetic code
If you're visiting from Tangled Bank #62 you've probably already noticed that this isn't your usual science blog. I rarely post about cool science from other sources, but concentrate on the research going on in my own lab. We're trying to understand how and why bacteria take up DNA (Is it food? Is it sex?), using a mix of molecular and bioinformatics approaches. I hope the things I write give others a peek into how research is really done, but I don't expect the day-to-day stuff to be of much interest to anyone except the other members of our lab (the sidebar has links to their blogs).

But sometimes I post about other things, and today I have a puzzle. The scintillation counter we use identifies its users' preset programs by number, and each job's program is flagged by a small card whose black-and-white pattern encodes the user number. The other day card #9 went missing, so we had to decipher the code so we could create a fake card #9.
Unfortunately other cards were also missing, but we were able to infer the pattern for card #9 from the five cards shown on the left. If you get stuck, there's a hint on the USS-R-US blog (link in the sidebar).
The rule we inferred didn't seem to make sense for user cards with numbers higher than 10; I'll post those patterns tomorrow.

But sometimes I post about other things, and today I have a puzzle. The scintillation counter we use identifies its users' preset programs by number, and each job's program is flagged by a small card whose black-and-white pattern encodes the user number. The other day card #9 went missing, so we had to decipher the code so we could create a fake card #9.
Unfortunately other cards were also missing, but we were able to infer the pattern for card #9 from the five cards shown on the left. If you get stuck, there's a hint on the USS-R-US blog (link in the sidebar).
The rule we inferred didn't seem to make sense for user cards with numbers higher than 10; I'll post those patterns tomorrow.
Mutants R US
My transformations worked, and I have thousands of what should be sxy purR double mutants. In fact I have thousands of double mutants for each of our five hypercompetence mutations.
Today I'll pick a single colony of each (starting from single colonies is the first rule of hygiene in bacterial genetics), and inoculate them so I can check that the mutants still are hypercompetent before I freeze them as lab stocks (checking strains before freezing them is the second rule of hygiene in bacterial genetics). I'll also streak them onto fresh kanamycin plates to give to the grad student who'll be testing the nucleotide effect.
And I just discovered that today it's my turn to do lab meeting! (I should have checked the schedule.) Luckily it's not until 3:30, and the posts I've been writing here should help me get my ideas organized.

1. Zinc test
2. purR test
3. motif searches
(The comic is from LabBratz.)
Today I'll pick a single colony of each (starting from single colonies is the first rule of hygiene in bacterial genetics), and inoculate them so I can check that the mutants still are hypercompetent before I freeze them as lab stocks (checking strains before freezing them is the second rule of hygiene in bacterial genetics). I'll also streak them onto fresh kanamycin plates to give to the grad student who'll be testing the nucleotide effect.
And I just discovered that today it's my turn to do lab meeting! (I should have checked the schedule.) Luckily it's not until 3:30, and the posts I've been writing here should help me get my ideas organized.

1. Zinc test
2. purR test
3. motif searches
(The comic is from LabBratz.)
Little steps
The purR knockout cells grew on the kanamycin agar, and I purified DNA from them. Today I'll grow up the hypercompetent mutants, give them some of the DNA, and plate them on kanamycin. These mutants are so competent that I won't need to starve them, and I don't even need to bother measuring the DNA concentration before I use it. If nothing goes wrong, tomorrow I should have my mutants.
Of course something usually does go wrong:
Of course something usually does go wrong:
"Most scientists spend most of their time trying to figure out why their experiments won't work."I spent most of the weekend on the motif-search program, alternately testing the effects of different inputs and trying to understand the brief manual. The 'read-me' included a list of input 'flags' that would change the program settings, but this was very terse and I alternated between discovering that I had the flag format wrong and suspecting that the flag was being ignored. The manual is quite dense with information. I've made about eight attempts to read it, each time making sense of a few more points, but I still feel a bit too ignorant/confused to be able to ask the authors sensible questions by email.R. Redfield
Maybe PurR does repress a competence gene after all
In a post last month I wondered whether the purine repressor PurR might repress competence genes, and concluded that this was probably a dead horse that I should stop flogging. But one of the grad students has some new results that suggest I was wrong.
We've known for years that adding purine nucleotides to the competence-inducing medium prevents transformation. (Oops, I just discovered that my sidebar link to the paper doesn't work any more, probably because we redid our lab web pages... ... Fixed.) I originally hypothesized that this was probably because the PurR repressor repressed competence genes, but a previous grad student knocked out the purR gene and showed that this didn't increase transformation (my hypothesis predicted it would). So I discarded the hypothesis.
Instead I hypothesized that the nucleotide effect was due to an effect of nucleotide pools on expression of the competence-gene activator Sxy. The grad student working on Sxy and CRP has now carefully examined the effect of nucleotides on sxy expression, measuring three things from each cell sample: Sxy protein, sxy mRNA, and transformation frequency. Consistent with this hypothesis, all were dramatically reduced by addition of nucleotides. So far so good.
We don't know how the purine nucleotides repress sxy, but we have a clue. I've probably mentioned before that we have mutations in the sxy gene that turn it on under conditions that normally keep it turned off. This suggested a new hypothesis, that the effect of these hypercompetence mutations is to make sxy expression independent of purine nucleotides (i.e. that the mutations negate the nucleotide effect).
The grad student has now tested this by examining the same three things (Sxy protein, sxy mRNA, and transformation frequency) from wild type and mutant cells treated with nucleotides. As predicted, addition of nucleotides to the mutants does NOT reduce sxy mRNA or Sxy protein. But, the mutants nevertheless show the same reduced transformation frequency as wild type cells.
Hmm... Sxy normally turns on all the competence genes, so why aren't the cells transforming? (Reminder: competence refers to uptake of DNA, and transformation to a genetic change caused by this DNA recombining with the cell's chromosome.) Could it be that not all of the competence genes are being turned on when Sxy and nucleotides are both present?
Back to PurR, which represses some (non-competence) genes when purines are high. The mutant results would make sense if one or more of the genes needed for transformation is repressed by PurR. We can use the purR knockout mutation to test this! All we need to do is to combine the purR mutation with one of the sxy hypercompetent mutations. If the hypothesis is correct, the transformation frequency of this double mutant should not be reduced by nucleotides.
So my job is to make the purR sxy double mutant, and the grad student will test its competence. My plan is to make (or find) some DNA of the purR mutant and transform it into one or more of the sxy hypercompetence mutants (we have five). Dead easy.
But there's a hitch. The grad student who made the purR mutant is long gone - he failed his comprehensive exams and left no forwarding address. I have his notebook but can't find his stock boxes in the fridges or freezers. Our lab freezer contains vials of what should be the purR mutant cells, but due to a freezer meltdown a couple of years ago most of these cells are probably dead. The mutant cells should be resistant to kanamycin, but my first attempt to grow cells from this stock produced no colonies on the kanamycin plate and only a few on a no-drug plate; I restreaked those yesterday and will find out today whether they are resistant. If not, I have backup plans.
We've known for years that adding purine nucleotides to the competence-inducing medium prevents transformation. (Oops, I just discovered that my sidebar link to the paper doesn't work any more, probably because we redid our lab web pages... ... Fixed.) I originally hypothesized that this was probably because the PurR repressor repressed competence genes, but a previous grad student knocked out the purR gene and showed that this didn't increase transformation (my hypothesis predicted it would). So I discarded the hypothesis.
Instead I hypothesized that the nucleotide effect was due to an effect of nucleotide pools on expression of the competence-gene activator Sxy. The grad student working on Sxy and CRP has now carefully examined the effect of nucleotides on sxy expression, measuring three things from each cell sample: Sxy protein, sxy mRNA, and transformation frequency. Consistent with this hypothesis, all were dramatically reduced by addition of nucleotides. So far so good.
We don't know how the purine nucleotides repress sxy, but we have a clue. I've probably mentioned before that we have mutations in the sxy gene that turn it on under conditions that normally keep it turned off. This suggested a new hypothesis, that the effect of these hypercompetence mutations is to make sxy expression independent of purine nucleotides (i.e. that the mutations negate the nucleotide effect).
The grad student has now tested this by examining the same three things (Sxy protein, sxy mRNA, and transformation frequency) from wild type and mutant cells treated with nucleotides. As predicted, addition of nucleotides to the mutants does NOT reduce sxy mRNA or Sxy protein. But, the mutants nevertheless show the same reduced transformation frequency as wild type cells.
Hmm... Sxy normally turns on all the competence genes, so why aren't the cells transforming? (Reminder: competence refers to uptake of DNA, and transformation to a genetic change caused by this DNA recombining with the cell's chromosome.) Could it be that not all of the competence genes are being turned on when Sxy and nucleotides are both present?
Back to PurR, which represses some (non-competence) genes when purines are high. The mutant results would make sense if one or more of the genes needed for transformation is repressed by PurR. We can use the purR knockout mutation to test this! All we need to do is to combine the purR mutation with one of the sxy hypercompetent mutations. If the hypothesis is correct, the transformation frequency of this double mutant should not be reduced by nucleotides.
So my job is to make the purR sxy double mutant, and the grad student will test its competence. My plan is to make (or find) some DNA of the purR mutant and transform it into one or more of the sxy hypercompetence mutants (we have five). Dead easy.
But there's a hitch. The grad student who made the purR mutant is long gone - he failed his comprehensive exams and left no forwarding address. I have his notebook but can't find his stock boxes in the fridges or freezers. Our lab freezer contains vials of what should be the purR mutant cells, but due to a freezer meltdown a couple of years ago most of these cells are probably dead. The mutant cells should be resistant to kanamycin, but my first attempt to grow cells from this stock produced no colonies on the kanamycin plate and only a few on a no-drug plate; I restreaked those yesterday and will find out today whether they are resistant. If not, I have backup plans.
Sneaking around the segmentation fault....
Now we have our Perl script to chop the genome sequence into short-enough fragments, and we have the motif-search program running on the fast Westgrid server. So I've been trying to run motif searches on the whole genome. It sort-of works. Actually, it almost works great!
What works: First, it usually runs without quitting prematurely. Second, it produces what I think is the correct output. I say I think because I don't understand the statistical parts of the output. Third, this time I asked it to search the sequence for 2 motifs rather than 1, and even that seems to be working. Fourth, much of the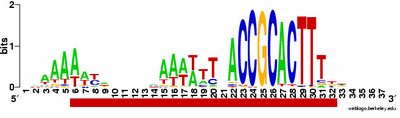 time the output shows the pattern I was expecting to see: alignment of hundreds of short sequences, each containing a sequence related to the previously characterized USS. I trim these down (using Word's search-and-destroy function) and paste them into WebLogo, which generates logos like that above, summarizing the pattern. And it's fast - analyzing the whole genome takes only 5-10 minutes.
time the output shows the pattern I was expecting to see: alignment of hundreds of short sequences, each containing a sequence related to the previously characterized USS. I trim these down (using Word's search-and-destroy function) and paste them into WebLogo, which generates logos like that above, summarizing the pattern. And it's fast - analyzing the whole genome takes only 5-10 minutes.
What isn't yet right: Sometimes it misses what should be the very significant motif and instead returns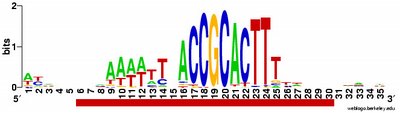 a weak motif that has nothing to do with the USS; I think this means I've set the stringency too low by telling it to expect too many sites with the motif. Often it returns only part of the USS motif, by cutting one side of the USS off, in favour of positions that show no evident similarity at all (when represented as WebLogos). This happens partly because it has decided not to fragment the motif into sub-motifs separated by non-consensus bases - I don't know why. The logo in this paragraph shows such a case. Compare it to the logo in the previous paragraph, and you see that the leftmost AT-rich part is missing. In both images the red underlining shows the positions that the motif search program decided had significant consensuses; in both the program has included positions with no consensuses and left out positions further to the left that would have strong consensuses. It could have included these positions by fragmenting the motif, but it didn't.
a weak motif that has nothing to do with the USS; I think this means I've set the stringency too low by telling it to expect too many sites with the motif. Often it returns only part of the USS motif, by cutting one side of the USS off, in favour of positions that show no evident similarity at all (when represented as WebLogos). This happens partly because it has decided not to fragment the motif into sub-motifs separated by non-consensus bases - I don't know why. The logo in this paragraph shows such a case. Compare it to the logo in the previous paragraph, and you see that the leftmost AT-rich part is missing. In both images the red underlining shows the positions that the motif search program decided had significant consensuses; in both the program has included positions with no consensuses and left out positions further to the left that would have strong consensuses. It could have included these positions by fragmenting the motif, but it didn't.
The biggest problem is the mysterious segmentation fault error. If it's using the full genome sequence (1.83megabases), and if I ask it to find a motif bigger than 18bp, the program begins the analysis but stops after a few cycles, reporting a segmentation fault. Googling segmentation fault tells me that this is probably because some string has become too long (the program is trying to put too much information into some location). I'm going to have to read the all-too-terse instructions to see if I can find a way around this. If I can't, I'm hoping that the person who sent me the binary code will take pity on my ignorance and help me solve the problem. The worst case will be if there is no way around this, but even then I think I can still get the analysis I need - it will just take more work on my part, combining results from different parts of the genome.
What works: First, it usually runs without quitting prematurely. Second, it produces what I think is the correct output. I say I think because I don't understand the statistical parts of the output. Third, this time I asked it to search the sequence for 2 motifs rather than 1, and even that seems to be working. Fourth, much of the
 time the output shows the pattern I was expecting to see: alignment of hundreds of short sequences, each containing a sequence related to the previously characterized USS. I trim these down (using Word's search-and-destroy function) and paste them into WebLogo, which generates logos like that above, summarizing the pattern. And it's fast - analyzing the whole genome takes only 5-10 minutes.
time the output shows the pattern I was expecting to see: alignment of hundreds of short sequences, each containing a sequence related to the previously characterized USS. I trim these down (using Word's search-and-destroy function) and paste them into WebLogo, which generates logos like that above, summarizing the pattern. And it's fast - analyzing the whole genome takes only 5-10 minutes.What isn't yet right: Sometimes it misses what should be the very significant motif and instead returns
 a weak motif that has nothing to do with the USS; I think this means I've set the stringency too low by telling it to expect too many sites with the motif. Often it returns only part of the USS motif, by cutting one side of the USS off, in favour of positions that show no evident similarity at all (when represented as WebLogos). This happens partly because it has decided not to fragment the motif into sub-motifs separated by non-consensus bases - I don't know why. The logo in this paragraph shows such a case. Compare it to the logo in the previous paragraph, and you see that the leftmost AT-rich part is missing. In both images the red underlining shows the positions that the motif search program decided had significant consensuses; in both the program has included positions with no consensuses and left out positions further to the left that would have strong consensuses. It could have included these positions by fragmenting the motif, but it didn't.
a weak motif that has nothing to do with the USS; I think this means I've set the stringency too low by telling it to expect too many sites with the motif. Often it returns only part of the USS motif, by cutting one side of the USS off, in favour of positions that show no evident similarity at all (when represented as WebLogos). This happens partly because it has decided not to fragment the motif into sub-motifs separated by non-consensus bases - I don't know why. The logo in this paragraph shows such a case. Compare it to the logo in the previous paragraph, and you see that the leftmost AT-rich part is missing. In both images the red underlining shows the positions that the motif search program decided had significant consensuses; in both the program has included positions with no consensuses and left out positions further to the left that would have strong consensuses. It could have included these positions by fragmenting the motif, but it didn't.The biggest problem is the mysterious segmentation fault error. If it's using the full genome sequence (1.83megabases), and if I ask it to find a motif bigger than 18bp, the program begins the analysis but stops after a few cycles, reporting a segmentation fault. Googling segmentation fault tells me that this is probably because some string has become too long (the program is trying to put too much information into some location). I'm going to have to read the all-too-terse instructions to see if I can find a way around this. If I can't, I'm hoping that the person who sent me the binary code will take pity on my ignorance and help me solve the problem. The worst case will be if there is no way around this, but even then I think I can still get the analysis I need - it will just take more work on my part, combining results from different parts of the genome.
How hard can it be?
Sunday evening I sent the post-doc who's been learning Perl an email asking if we could write a little 'script' that would take a long DNA sequence and break it into shorter fragments of a specified length. (Short Perl programs are often called 'scripts'; I suspect this may be a more general term for program modules.) The sequences need to be in a style called 'FASTA format', which requires each sequence to start on a new line, and to be preceded by a line starting with ">". Even with our very limited Perl skills I figured this should only take us a little while.
I need this done because the 'unbiased motif search' program I'm using to look at G-USSs (or whatever we're calling them) in the genome will only accept sequences that are ≤10,000bp long. The genome is about 1,830,000bp long, and I didn't want to have to go through it by hand finding the right places to insert carriage returns and ">"s. I'm also starting to think we should do similar motif searches on lots of other genomes, so having this process automated would be a big help.
This afternoon we (the post-doc, mostly) finally got it to work. The problem wasn't so much our lack of Perl skills as our realization that we had to include other steps that would clean up the sequence before it could be used.
For example the motif-search program rejects any sequences containing letters other than A, C, G, T and N. A, C, G and T are the bases in DNA, and N is used when we're not sure what base is present at a particular position. The sequences we're using include other letters indicating other kinds of uncertainties; N means "could be any of A, C, G or T', W means "could be A or T", Y means "could be C or T", etc. So we needed our script to go through the sequence, find all the Ys and Ws (and Ks and Rs and Ss and Ms) and replace them with Ns.
Then we realized that we also had to get rid of any line feeds (end-of-line characters) in the input sequence. These can be invisible in the sequence but will send Perl into a tailspin, especially if they're the peculiar line feeds that Macs use. We finally gave up on trying to get Perl to remove them, but found an easy way to have Unix remove them from the sequence before we give it to the Perl script.
One big obstacle was getting the script to insert the ">" breaks where we wanted them. Our strategy was to have the script divide the base number by the interval at which we wanted the breaks, and only put a break when this remainder equaled zero. (example: Say the desired interval is 500bp. Only at positions 500, 1000, 1500, 2000 etc. will the remainder when divided by 500 = 0.) Coding this should have been easy, because algorithms for calculating remainders are built into Perl. But either we had the instructions for calculating the remainder backwards, or we just got 'divisor' and 'dividend' confused (hey, grade 4 was a long time ago).
But now it works.
I think I also discovered why I wasn't getting responses from the in-line interface to the motif search software. RTFMing (RingTFM?) revealed that I had been putting the wrong kind of numbers into a couple of the boxes. Because the responses from this interface usually take a day to arrive (by email) I won't know until tomorrow whether that was really the problem - I know it was A problem, but I don't know if it was the reason I got no responses, as I also got no error messages.
The post-doc also may have succeeded in getting a version of the motif-search program running just for us on the whiz-bang computer system WestGrid. It will be a bit more cumbersome to set up than the online version, but we should be able to get lots of results fast.
Added the next day: The guy in the next office just showed me some bioinformatics software that, among other things, does just what our new Perl script does. Oh well, we're still proud of having done it ourselves.
I need this done because the 'unbiased motif search' program I'm using to look at G-USSs (or whatever we're calling them) in the genome will only accept sequences that are ≤10,000bp long. The genome is about 1,830,000bp long, and I didn't want to have to go through it by hand finding the right places to insert carriage returns and ">"s. I'm also starting to think we should do similar motif searches on lots of other genomes, so having this process automated would be a big help.
This afternoon we (the post-doc, mostly) finally got it to work. The problem wasn't so much our lack of Perl skills as our realization that we had to include other steps that would clean up the sequence before it could be used.
For example the motif-search program rejects any sequences containing letters other than A, C, G, T and N. A, C, G and T are the bases in DNA, and N is used when we're not sure what base is present at a particular position. The sequences we're using include other letters indicating other kinds of uncertainties; N means "could be any of A, C, G or T', W means "could be A or T", Y means "could be C or T", etc. So we needed our script to go through the sequence, find all the Ys and Ws (and Ks and Rs and Ss and Ms) and replace them with Ns.
Then we realized that we also had to get rid of any line feeds (end-of-line characters) in the input sequence. These can be invisible in the sequence but will send Perl into a tailspin, especially if they're the peculiar line feeds that Macs use. We finally gave up on trying to get Perl to remove them, but found an easy way to have Unix remove them from the sequence before we give it to the Perl script.
One big obstacle was getting the script to insert the ">" breaks where we wanted them. Our strategy was to have the script divide the base number by the interval at which we wanted the breaks, and only put a break when this remainder equaled zero. (example: Say the desired interval is 500bp. Only at positions 500, 1000, 1500, 2000 etc. will the remainder when divided by 500 = 0.) Coding this should have been easy, because algorithms for calculating remainders are built into Perl. But either we had the instructions for calculating the remainder backwards, or we just got 'divisor' and 'dividend' confused (hey, grade 4 was a long time ago).
But now it works.
I think I also discovered why I wasn't getting responses from the in-line interface to the motif search software. RTFMing (RingTFM?) revealed that I had been putting the wrong kind of numbers into a couple of the boxes. Because the responses from this interface usually take a day to arrive (by email) I won't know until tomorrow whether that was really the problem - I know it was A problem, but I don't know if it was the reason I got no responses, as I also got no error messages.
The post-doc also may have succeeded in getting a version of the motif-search program running just for us on the whiz-bang computer system WestGrid. It will be a bit more cumbersome to set up than the online version, but we should be able to get lots of results fast.
Added the next day: The guy in the next office just showed me some bioinformatics software that, among other things, does just what our new Perl script does. Oh well, we're still proud of having done it ourselves.
G-USSs and R-USSs (C-USSs and U-USSs?)
One of the projects we're working on will produce a paper titled "Defining the Haemophilus influenzae uptake signal sequence". Today I realized that a key conceptual step is making a clear distinction between two ways we've been using the term USS.
First, we've been using it to describe the sequence preference of the cell-surface machinery that takes up DNA. Second, we've been using it to describe the consensus of the many repeats in the cell's genome that appear to match this preference. The similarity between the preference and the repeats is much too strong to be a coincidence, but that doesn't justify confounding the two meanings, as we and others have done.
In previous papers (ours and others) the preference has been used as a guide to investigating the consensus, and vice versa. But in the present project we aim to define them separately. The first part of the project is the 'unbiased' analysis of USS-like repeats in the genome I've posted about previously. The second part will be a reanalysis of old uptake data (see New bottles for old wine) and the third will be direct tests of how changing different positions in the sequence changes uptake.
We would like to create terms for the two kinds of "USS"s: the preference and the consensus. I started with "USS" for the preference and "USS-like sequence" for the consensus. But this suggests that the consensus isn't the same as the preference, though our null hypothesis predicts it will be. "USS-related sequence" isn't much better. Today at lab meeting one of the grad students suggested using a letter in front of "USS' to indicate which we are referring to: "c-USS" (or "g-USS") for the consensus in the chromosome (or genome), and "r-USS" for the preference of the receptor on the cell surface.
We expect our results to indicate that the preference and the consensus are the same sequence. If so these terms will never need to be used outside of this paper, and it won't matter much if they are ugly and/or cumbersome. But on the off chance that the two sequences turn out to be different, we should at least try to find some terms we can live with.
First, we've been using it to describe the sequence preference of the cell-surface machinery that takes up DNA. Second, we've been using it to describe the consensus of the many repeats in the cell's genome that appear to match this preference. The similarity between the preference and the repeats is much too strong to be a coincidence, but that doesn't justify confounding the two meanings, as we and others have done.
In previous papers (ours and others) the preference has been used as a guide to investigating the consensus, and vice versa. But in the present project we aim to define them separately. The first part of the project is the 'unbiased' analysis of USS-like repeats in the genome I've posted about previously. The second part will be a reanalysis of old uptake data (see New bottles for old wine) and the third will be direct tests of how changing different positions in the sequence changes uptake.
We would like to create terms for the two kinds of "USS"s: the preference and the consensus. I started with "USS" for the preference and "USS-like sequence" for the consensus. But this suggests that the consensus isn't the same as the preference, though our null hypothesis predicts it will be. "USS-related sequence" isn't much better. Today at lab meeting one of the grad students suggested using a letter in front of "USS' to indicate which we are referring to: "c-USS" (or "g-USS") for the consensus in the chromosome (or genome), and "r-USS" for the preference of the receptor on the cell surface.
We expect our results to indicate that the preference and the consensus are the same sequence. If so these terms will never need to be used outside of this paper, and it won't matter much if they are ugly and/or cumbersome. But on the off chance that the two sequences turn out to be different, we should at least try to find some terms we can live with.
Still looking for equilibrium
Today I thought more about an algorithm (a rule, a test, a way to calculate) I'd outlined that would decide when our Perl simulations of USS accumulation had reached equilibrium. (I had to think more because my initial explanation was very hard to follow.)
It's actually two algorithms, one for deciding how often to report the latest status of the simulated genome, and one to decide whether, at a report time, equilibrium has been reached and the run should stop.
The first algorithm is needed because how many cycles a run takes depends very strongly on the conditions we set. We want frequent reports, but if the run is going to take 500,000 cycles we don't want a report every cycle, or even every 10 or 100 cycles. How often we want reports also changes within a single run: at the start of the run things are changing fast and we want frequent reports, but as conditions approach equilibrium we want reports less often.
Here the solution is to make the reporting frequency a function of how many cycles have elapsed. The new rule is: only report results if more than 1% of the total run has elapsed since the last report. The result is that, for the first 100 cycles we get a report every cycle, but after 1000 cycles we only get a report every 10 cycles, and after 10,000 cycles we only get one every 100 cycles. Even if the run lasts 1,000,000 cycles, we'll only have about than 1000 reports. The best thing is we don't need to have any idea of how long the run might take.
The second algorithm is more complicated and I'm not going to describe it until we've had a chance to try it out.
It's actually two algorithms, one for deciding how often to report the latest status of the simulated genome, and one to decide whether, at a report time, equilibrium has been reached and the run should stop.
The first algorithm is needed because how many cycles a run takes depends very strongly on the conditions we set. We want frequent reports, but if the run is going to take 500,000 cycles we don't want a report every cycle, or even every 10 or 100 cycles. How often we want reports also changes within a single run: at the start of the run things are changing fast and we want frequent reports, but as conditions approach equilibrium we want reports less often.
Here the solution is to make the reporting frequency a function of how many cycles have elapsed. The new rule is: only report results if more than 1% of the total run has elapsed since the last report. The result is that, for the first 100 cycles we get a report every cycle, but after 1000 cycles we only get a report every 10 cycles, and after 10,000 cycles we only get one every 100 cycles. Even if the run lasts 1,000,000 cycles, we'll only have about than 1000 reports. The best thing is we don't need to have any idea of how long the run might take.
The second algorithm is more complicated and I'm not going to describe it until we've had a chance to try it out.
Is the origin of biological chirality a no-brainer?
One argument invoked by creationists for a magical origin of life is that the observed homochirality (definition below) of biological molecules could not have arisen otherwise. Creationists are notorious for their sloppy reasoning, but I'm a bit puzzled that scientists also see this as a big problem. Here's an example from the Answers in Genesis people, and the blue box in this article is an example from PLoS Biology.
The fundamental argument seems to be that some special forces or factors are needed to explain how and/or why the first living things used only D-sugars and only L-amino acids. It's often claimed that this would not have happened unless the starting abiotic materials had an excess of one enantiomer (definition below) over the other.
I think people have fallen into the error of assuming that, at the molecular level, enantiomers are much more similar to each other than to other related molecules. But I don't think it's really any harder for an asymmetric reactant or catalyst to distinguish D-glucose from L-glucose than from either enantiomer of fructose or galactose. Or to distinguish L-leucine from D-leucine than from D- or L-isoleucine. They may contain the same atoms but they all have entirely different shapes, and so they are all entirely different molecules.
(Here's the same point made about words. The words pacer and recap are palindromes, but most readers have no more trouble telling them apart than telling either from caper.)
Chemists discovered chirality when investigating the bulk properties of pure crystals and solutions. Chemical synthesis of a chiral molecule from simpler non-chiral precursors usually produces equal amounts of both enantiomers, whose identical physical properties make them very difficult to separate. Chemists thus view enantiomers as almost-identical molecules, differing only in their 'handedness'.
But chemists are late arrivals on the evolutionary scene, and the first self-replicating entities would usually have interacted with individual molecules in complex mixtures. Because all but the simplest of biologically relevant molecules are asymmetric, most inter-molecular interactions would always have been between asymmetric participants, each no more likely to confuse their partner with its enantiomer than with any other molecule. The fact that crystals of D-glucose and L-glucose have the same bulk properties (solubility, melting temperature) would have been irrelevant.
We don't need to fuss with defining 'life', but can simply think about the origin of entities capable of evolving by natural selection (having heritable variation causing differential reproduction). Any molecule complex enough to have heritable variation would certainly have been complex enough to be asymmetric. To such molecules, discrimination between enantiomers wouldn't have been any more of a problem than discriminating between other possible reactants.
Although this now seems so obvious to me, I only realized it a couple of years ago. Until then I unquestioningly accepted the general teaching that the origin of biological chirality was a big problem. Am I missing something?
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Definitions of enantiomer and homochirality: Like letters in words, the same combinations of atoms can be put together in different arrangements. Sometimes the arrangements can be mirror-images of each other. Chemists call molecules that contain the same atoms connected in mirror-symmetry arrangements enantiomers of each other. Enantiomers are distinct molecules (like the words speed and deeps), but pure preparations of these chiral molecules have the same bulk physical properties. The molecules found in organisms are usually homochiral: only one of the two enantiomers is used. Our metabolism uses only the D enantiomers of sugars and only the L enantiomers of amino acids.
The fundamental argument seems to be that some special forces or factors are needed to explain how and/or why the first living things used only D-sugars and only L-amino acids. It's often claimed that this would not have happened unless the starting abiotic materials had an excess of one enantiomer (definition below) over the other.
I think people have fallen into the error of assuming that, at the molecular level, enantiomers are much more similar to each other than to other related molecules. But I don't think it's really any harder for an asymmetric reactant or catalyst to distinguish D-glucose from L-glucose than from either enantiomer of fructose or galactose. Or to distinguish L-leucine from D-leucine than from D- or L-isoleucine. They may contain the same atoms but they all have entirely different shapes, and so they are all entirely different molecules.
(Here's the same point made about words. The words pacer and recap are palindromes, but most readers have no more trouble telling them apart than telling either from caper.)
Chemists discovered chirality when investigating the bulk properties of pure crystals and solutions. Chemical synthesis of a chiral molecule from simpler non-chiral precursors usually produces equal amounts of both enantiomers, whose identical physical properties make them very difficult to separate. Chemists thus view enantiomers as almost-identical molecules, differing only in their 'handedness'.
But chemists are late arrivals on the evolutionary scene, and the first self-replicating entities would usually have interacted with individual molecules in complex mixtures. Because all but the simplest of biologically relevant molecules are asymmetric, most inter-molecular interactions would always have been between asymmetric participants, each no more likely to confuse their partner with its enantiomer than with any other molecule. The fact that crystals of D-glucose and L-glucose have the same bulk properties (solubility, melting temperature) would have been irrelevant.
We don't need to fuss with defining 'life', but can simply think about the origin of entities capable of evolving by natural selection (having heritable variation causing differential reproduction). Any molecule complex enough to have heritable variation would certainly have been complex enough to be asymmetric. To such molecules, discrimination between enantiomers wouldn't have been any more of a problem than discriminating between other possible reactants.
Although this now seems so obvious to me, I only realized it a couple of years ago. Until then I unquestioningly accepted the general teaching that the origin of biological chirality was a big problem. Am I missing something?
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Definitions of enantiomer and homochirality: Like letters in words, the same combinations of atoms can be put together in different arrangements. Sometimes the arrangements can be mirror-images of each other. Chemists call molecules that contain the same atoms connected in mirror-symmetry arrangements enantiomers of each other. Enantiomers are distinct molecules (like the words speed and deeps), but pure preparations of these chiral molecules have the same bulk physical properties. The molecules found in organisms are usually homochiral: only one of the two enantiomers is used. Our metabolism uses only the D enantiomers of sugars and only the L enantiomers of amino acids.
Not zinc
OK, the results are in, and zinc doesn't help cells take up DNA. Here's the data:
Adding zinc doesn't increase transformation frequency (the simplest and most sensitive measure of DNA uptake). The small differences between most these columns aren't significant, given the modest scale of this experiment. High concentrations of zinc are probably a bit toxic: at 1mM zinc decreased transformation more than 2-fold, and at 3mM it decreased cell viability 2-fold. (I missed adding the DNA to the 3mM tube so I don't have this transformation frequency.) Adding the zinc chelator histidine didn't decrease DNA uptake either.
The conclusion: we can probably rule out an important role for zinc or zinc-finger proteins in DNA uptake. Is this an important enough experiment that I should do it over several times, to get data with good error bars? It's an important experiment, but if the possible effect of zinc is too small to have shown up in this experiment it's probably too small to be interesting.
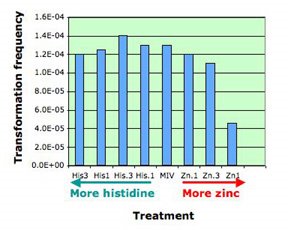
Adding zinc doesn't increase transformation frequency (the simplest and most sensitive measure of DNA uptake). The small differences between most these columns aren't significant, given the modest scale of this experiment. High concentrations of zinc are probably a bit toxic: at 1mM zinc decreased transformation more than 2-fold, and at 3mM it decreased cell viability 2-fold. (I missed adding the DNA to the 3mM tube so I don't have this transformation frequency.) Adding the zinc chelator histidine didn't decrease DNA uptake either.
The conclusion: we can probably rule out an important role for zinc or zinc-finger proteins in DNA uptake. Is this an important enough experiment that I should do it over several times, to get data with good error bars? It's an important experiment, but if the possible effect of zinc is too small to have shown up in this experiment it's probably too small to be interesting.
Lessons from the classics?
I've been reading the classics - papers on Haemophilus influenzae competence and transformation published in the 1960s. They contain lots of information that, if true, is important for our work. But we know it's all too easy to 'discover' things that turn out to not be true, so we'll need to proceed cautiously.
Here are some factoids that contradict my current assumptions:
The media additive hemin loses potency after for a few weeks in the fridge . (We've been keeping it for years.)
Washing competent cells removes one or more factors needed for DNA uptake.
Cells have a nucleoside phosphorylase that is active in the periplasm - this means it acts on nucleosides that haven't been taken up by the cell yet. This may be important for understanding why purine nucleotides and nucleosides inhibit competence but purine bases don't.
Cells bind about five times more DNA than they take up.
Here are some factoids that contradict my current assumptions:
The media additive hemin loses potency after for a few weeks in the fridge . (We've been keeping it for years.)
Washing competent cells removes one or more factors needed for DNA uptake.
Cells have a nucleoside phosphorylase that is active in the periplasm - this means it acts on nucleosides that haven't been taken up by the cell yet. This may be important for understanding why purine nucleotides and nucleosides inhibit competence but purine bases don't.
Cells bind about five times more DNA than they take up.
Zinc!
Zinc? I just realized that we've been neglecting the possible role in DNA uptake of divalent cations, especially zinc.
There are lots of reasons why we shouldn't have. A paper discussing DNA's ability to deform under stress showed that addition of zinc allowed kinks to form. There's a whole family of DNA-binding proteins called zinc finger proteins, because zinc mediates an interaction between histidines in the protein and DNA. Some of the ancient (45 years old) classic papers about competence point out that divalent cations influence DNA uptake. And every beginning biochemist learns that divalent cations help proteins interact with DNA.
The standard medium for inducing competence (which we also use for assaying DNA uptake and transformation), contains Mg++ and Ca++. The ingredients list doesn't include zinc, but it may be present as a contaminant, and variation in the level of contamination might even explain the long-term variations we see in transformation frequency. And it should be easy to test - just supplement the usual DNA-uptake solution with different amounts of a zinc salt or the zinc-chelating amino acid histidine. We have a couple of grams of histidine on the shelf, and for some reason a whole kg of ZnSO4. And it's high time I got back into the lab and did an experiment.
Experiment plan for tomorrow:
I think I have lots of competent cells in the freezer. Oops, no I don't, so I will need to grow some wildtype cells and make them competent. We have lots of DNA with a gene giving resistance to the antibiotic novobiocin, to use for the transformations. I'll need to make some agar plates, both plain sBHI (the usual nutrients) and with novobiocin added to select for my transformants.
I'll need to make up solutions of ZnSO4 (50 ml of 100mM) and histidine (small volume of 100mM?) and filter-sterilize them. The zinc-kink paper used 1mM Zn, but said 0.1mM worked too, so I'll try both. Maybe I should try a wide range of values, in case too much zinc is as bad as too little. The standard competence solution contains a little bit of histidine (0.013mg/ml - at 210 grams/mole that's 0.062mM). I'll also try adding histidine at 0.1mM and 1mM.
If zinc does help cells take up DNA, I should see higher transformation frequencies when the uptake solution is supplemented with zinc, and/or lower transformation frequencies when it is supplemented with histidine. The 'and/or' is because I don't know whether the uptake solution already contains significant amounts of zinc.
This will be fine old-fashioned microbiology experiment, and I'm pretty sure I can do it in one day. It's the weekend so there shouldn't be a lot of interuptions.
There are lots of reasons why we shouldn't have. A paper discussing DNA's ability to deform under stress showed that addition of zinc allowed kinks to form. There's a whole family of DNA-binding proteins called zinc finger proteins, because zinc mediates an interaction between histidines in the protein and DNA. Some of the ancient (45 years old) classic papers about competence point out that divalent cations influence DNA uptake. And every beginning biochemist learns that divalent cations help proteins interact with DNA.
The standard medium for inducing competence (which we also use for assaying DNA uptake and transformation), contains Mg++ and Ca++. The ingredients list doesn't include zinc, but it may be present as a contaminant, and variation in the level of contamination might even explain the long-term variations we see in transformation frequency. And it should be easy to test - just supplement the usual DNA-uptake solution with different amounts of a zinc salt or the zinc-chelating amino acid histidine. We have a couple of grams of histidine on the shelf, and for some reason a whole kg of ZnSO4. And it's high time I got back into the lab and did an experiment.
Experiment plan for tomorrow:
I think I have lots of competent cells in the freezer. Oops, no I don't, so I will need to grow some wildtype cells and make them competent. We have lots of DNA with a gene giving resistance to the antibiotic novobiocin, to use for the transformations. I'll need to make some agar plates, both plain sBHI (the usual nutrients) and with novobiocin added to select for my transformants.
I'll need to make up solutions of ZnSO4 (50 ml of 100mM) and histidine (small volume of 100mM?) and filter-sterilize them. The zinc-kink paper used 1mM Zn, but said 0.1mM worked too, so I'll try both. Maybe I should try a wide range of values, in case too much zinc is as bad as too little. The standard competence solution contains a little bit of histidine (0.013mg/ml - at 210 grams/mole that's 0.062mM). I'll also try adding histidine at 0.1mM and 1mM.
If zinc does help cells take up DNA, I should see higher transformation frequencies when the uptake solution is supplemented with zinc, and/or lower transformation frequencies when it is supplemented with histidine. The 'and/or' is because I don't know whether the uptake solution already contains significant amounts of zinc.
This will be fine old-fashioned microbiology experiment, and I'm pretty sure I can do it in one day. It's the weekend so there shouldn't be a lot of interuptions.
Is the USS kinky?
I've mentioned that I think the DNA-uptake process must start with creating a kink in the DNA at the USS sequence, before it can be transported across the outer membrane. Why do I think that, and what are the implications?
'Gram negative' bacterial cells such as H. influenzae have two membranes enclosing their cytoplasm, and DNA must travel across both of these and the intervening layer of periplasm and mesh-like cell wall. Here I'll be talking mainly about uptake across the H. influenzae outer membrane.
Transport of DNA across this or any membrane is predicted to be difficult because the membrane is hydrophobic and DNA is hydrophilic. That is, the membrane is made of lipids and repels charged molecules including water, whereas DNA has lots of negative charge and is surrounded by water and positively charged ions. Thus DNA and membranes will repel each other.
Because lots of valuable nutrients are hydrophilic, cells have evolved protein-lined pores in their membranes through which particular charged molecules can pass. But DNA presents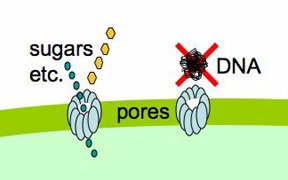 special problems, because it exists as very long stiff fibers, much much longer than the small sugar and amino acid molecules that most pores transport. Nevertheless, cells easily take up DNA molecules more than 50,000 base pairs long, even though these molecules are longer than the cells. A DNA fiber is very thin, and on the scale of a cell it's very flexible, so there's no problem fitting this much DNA into the cell. In fact H. influenzae cells already contain about 40 times this much DNA in their own chromosomes.
special problems, because it exists as very long stiff fibers, much much longer than the small sugar and amino acid molecules that most pores transport. Nevertheless, cells easily take up DNA molecules more than 50,000 base pairs long, even though these molecules are longer than the cells. A DNA fiber is very thin, and on the scale of a cell it's very flexible, so there's no problem fitting this much DNA into the cell. In fact H. influenzae cells already contain about 40 times this much DNA in their own chromosomes.
DNA uptake is problematic because, on a molecular scale, DNA is quite stiff. Its stiffness is described by its 'persistence length', which is about 50nm. As typical pores are only a few nm across, this means that DNA won't spontaneously collapse into a clump small enough that can pass through such a pore (like the DNA shown above with a big red X).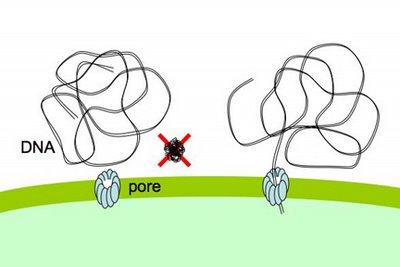
The DNA fiber is only about 2nm across, so its stiffness wouldn't be a problem if it could be 'threaded' through a pore, starting from one end. Work mainly from other labs strongly suggests that the channel is formed by the same proteins that form the channel used by a protein filament, the type 4 pilus, which cells can extend and retract from their surface. These channels are about 6nm across, more than wide enough for DNA to thread through.
But I suspect that the uptake machinery doesn't need to find the DNA's end in order to take it up. That is, I don't think DNA is usually threaded through the pilin channel.
The evidence for end-independent DNA uptake is that competent cells have no trouble transporting circular plasmids across the outer membrane, and that the plasmids remain unbroken in the periplasm (though they can't be transported across the inner membrane without being cut or broken). I need to reevaluate the strength of the evidence that the plasmid DNA doesn't get broken, but for now let's assume that it's solid.
How then does the plasmid get across the membrane? I propose that the DNA is sharply bent or kinked by the uptake machinery, allowing it to be poked in through the pilin channel. I say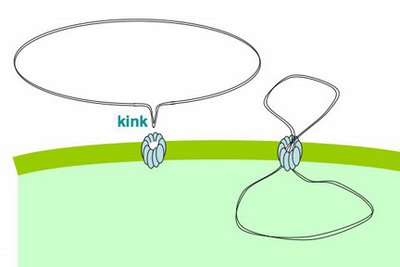 'by the uptake machinery' because normal DNA doesn't spontaneously form kinks, though it can be a bit curved. I also propose that one reason for preferential uptake of DNA with USS sequences is that these sequences are particularly easy to bend or kink. Once the kinked DNA is poked through the channel, the rest of it can be pulled in without additional kinking. This hypothetical process is illustrated in the little movie on our lab home page. The movie also shows the end of the DNA being pulled into the periplasm, and one strand of the DNA then being transported across the inner membrane.
'by the uptake machinery' because normal DNA doesn't spontaneously form kinks, though it can be a bit curved. I also propose that one reason for preferential uptake of DNA with USS sequences is that these sequences are particularly easy to bend or kink. Once the kinked DNA is poked through the channel, the rest of it can be pulled in without additional kinking. This hypothetical process is illustrated in the little movie on our lab home page. The movie also shows the end of the DNA being pulled into the periplasm, and one strand of the DNA then being transported across the inner membrane.
Unfortunately, I don't have any direct evidence that DNA with the USS sequence is any easier to kink than other DNA.
One way to create a kink would be to separate the two DNA strands (single strands are much more flexible than a double-helix). The AT-rich sequences on one side of the USS core are good candidates for this kind of kinking, because their weak base pairs are more easily separated than GC base pairs. But I have only a very naive understanding of how this and other forces interact to determine whether a given DNA sequence can easily be kinked. I think it's time to consult an expert.
'Gram negative' bacterial cells such as H. influenzae have two membranes enclosing their cytoplasm, and DNA must travel across both of these and the intervening layer of periplasm and mesh-like cell wall. Here I'll be talking mainly about uptake across the H. influenzae outer membrane.
Transport of DNA across this or any membrane is predicted to be difficult because the membrane is hydrophobic and DNA is hydrophilic. That is, the membrane is made of lipids and repels charged molecules including water, whereas DNA has lots of negative charge and is surrounded by water and positively charged ions. Thus DNA and membranes will repel each other.
Because lots of valuable nutrients are hydrophilic, cells have evolved protein-lined pores in their membranes through which particular charged molecules can pass. But DNA presents
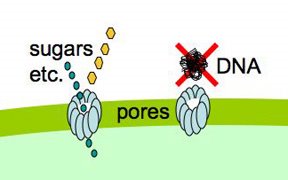 special problems, because it exists as very long stiff fibers, much much longer than the small sugar and amino acid molecules that most pores transport. Nevertheless, cells easily take up DNA molecules more than 50,000 base pairs long, even though these molecules are longer than the cells. A DNA fiber is very thin, and on the scale of a cell it's very flexible, so there's no problem fitting this much DNA into the cell. In fact H. influenzae cells already contain about 40 times this much DNA in their own chromosomes.
special problems, because it exists as very long stiff fibers, much much longer than the small sugar and amino acid molecules that most pores transport. Nevertheless, cells easily take up DNA molecules more than 50,000 base pairs long, even though these molecules are longer than the cells. A DNA fiber is very thin, and on the scale of a cell it's very flexible, so there's no problem fitting this much DNA into the cell. In fact H. influenzae cells already contain about 40 times this much DNA in their own chromosomes.DNA uptake is problematic because, on a molecular scale, DNA is quite stiff. Its stiffness is described by its 'persistence length', which is about 50nm. As typical pores are only a few nm across, this means that DNA won't spontaneously collapse into a clump small enough that can pass through such a pore (like the DNA shown above with a big red X).

The DNA fiber is only about 2nm across, so its stiffness wouldn't be a problem if it could be 'threaded' through a pore, starting from one end. Work mainly from other labs strongly suggests that the channel is formed by the same proteins that form the channel used by a protein filament, the type 4 pilus, which cells can extend and retract from their surface. These channels are about 6nm across, more than wide enough for DNA to thread through.
But I suspect that the uptake machinery doesn't need to find the DNA's end in order to take it up. That is, I don't think DNA is usually threaded through the pilin channel.
The evidence for end-independent DNA uptake is that competent cells have no trouble transporting circular plasmids across the outer membrane, and that the plasmids remain unbroken in the periplasm (though they can't be transported across the inner membrane without being cut or broken). I need to reevaluate the strength of the evidence that the plasmid DNA doesn't get broken, but for now let's assume that it's solid.
How then does the plasmid get across the membrane? I propose that the DNA is sharply bent or kinked by the uptake machinery, allowing it to be poked in through the pilin channel. I say
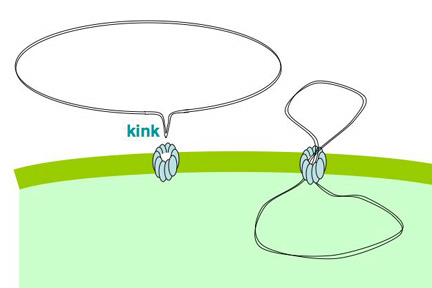 'by the uptake machinery' because normal DNA doesn't spontaneously form kinks, though it can be a bit curved. I also propose that one reason for preferential uptake of DNA with USS sequences is that these sequences are particularly easy to bend or kink. Once the kinked DNA is poked through the channel, the rest of it can be pulled in without additional kinking. This hypothetical process is illustrated in the little movie on our lab home page. The movie also shows the end of the DNA being pulled into the periplasm, and one strand of the DNA then being transported across the inner membrane.
'by the uptake machinery' because normal DNA doesn't spontaneously form kinks, though it can be a bit curved. I also propose that one reason for preferential uptake of DNA with USS sequences is that these sequences are particularly easy to bend or kink. Once the kinked DNA is poked through the channel, the rest of it can be pulled in without additional kinking. This hypothetical process is illustrated in the little movie on our lab home page. The movie also shows the end of the DNA being pulled into the periplasm, and one strand of the DNA then being transported across the inner membrane.Unfortunately, I don't have any direct evidence that DNA with the USS sequence is any easier to kink than other DNA.
One way to create a kink would be to separate the two DNA strands (single strands are much more flexible than a double-helix). The AT-rich sequences on one side of the USS core are good candidates for this kind of kinking, because their weak base pairs are more easily separated than GC base pairs. But I have only a very naive understanding of how this and other forces interact to determine whether a given DNA sequence can easily be kinked. I think it's time to consult an expert.
Representing a single USS
Previously I've posted about different ways to represent the consensus derived from comparing USS sequences. Lately I've been doing 'unbiased' searches that find USS-related repeats in parts of the H. influenzae genome, and visualizing their consensuses as SequenceLogos. I want to do this for the whole genome but I'm still working out how to give such a big sequence to the motif-search program. So far the unbiased USS consensuses I've made look very much like the 'biased' consensuses we've already derived. This result is a bit boring, but it's also reassuring. I'd hate to only now discover that we'd been using an inappropriate sequence for all our past work.
I continue to entertain the (so far unsupported) idea that USSs are sequences at which DNA can be easily kinked to permit its passage across the bacterial outer membrane. (The animation on our lab home page shows how I imagine this happening.) I've been concerned that thinking only about a USS consensus that's the average of all the real USS might mask some important properties of individual USSs, so I'm also trying to understand the properties of single real USS sequences.
I've just found a web site called MDDNA, which analyzes the predicted structure of any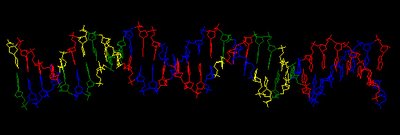 short DNA sequence you paste into it, using the latest predictions of how different combinations of neighbouring bases modulate the standard "B" structure of the DNA double helix. The image at the left shows the structure of a typical USS. This is just a screenshot - the MDDNA analysis creates a "PDB" structure file and lets you view and play with it in 3-D in a "Jmol" applet.
short DNA sequence you paste into it, using the latest predictions of how different combinations of neighbouring bases modulate the standard "B" structure of the DNA double helix. The image at the left shows the structure of a typical USS. This is just a screenshot - the MDDNA analysis creates a "PDB" structure file and lets you view and play with it in 3-D in a "Jmol" applet.
The site also provides all the 'helicoidal parameters' associated with the structure it has produced. But I'll need to do some work before I can understand what these mean.
You've probably noticed that the DNA in the image is not bent at all (certainly not sharply kinked as I suggested above). Neither were any of the molecules predicted from the other USS sequences I tried. This might mean that USS sequences are not prone to bending at all (i.e. my idea is wrong). Or it might be that the sequences are straight when free in solution, but bend easily when bound to their receptor (i.e. my idea could be right). I need to keep looking and reading to find out more about the dynamic properties of DNA molecules under stress.
I continue to entertain the (so far unsupported) idea that USSs are sequences at which DNA can be easily kinked to permit its passage across the bacterial outer membrane. (The animation on our lab home page shows how I imagine this happening.) I've been concerned that thinking only about a USS consensus that's the average of all the real USS might mask some important properties of individual USSs, so I'm also trying to understand the properties of single real USS sequences.
I've just found a web site called MDDNA, which analyzes the predicted structure of any
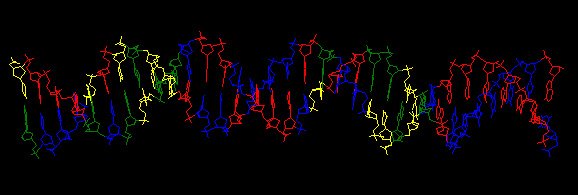 short DNA sequence you paste into it, using the latest predictions of how different combinations of neighbouring bases modulate the standard "B" structure of the DNA double helix. The image at the left shows the structure of a typical USS. This is just a screenshot - the MDDNA analysis creates a "PDB" structure file and lets you view and play with it in 3-D in a "Jmol" applet.
short DNA sequence you paste into it, using the latest predictions of how different combinations of neighbouring bases modulate the standard "B" structure of the DNA double helix. The image at the left shows the structure of a typical USS. This is just a screenshot - the MDDNA analysis creates a "PDB" structure file and lets you view and play with it in 3-D in a "Jmol" applet.The site also provides all the 'helicoidal parameters' associated with the structure it has produced. But I'll need to do some work before I can understand what these mean.
You've probably noticed that the DNA in the image is not bent at all (certainly not sharply kinked as I suggested above). Neither were any of the molecules predicted from the other USS sequences I tried. This might mean that USS sequences are not prone to bending at all (i.e. my idea is wrong). Or it might be that the sequences are straight when free in solution, but bend easily when bound to their receptor (i.e. my idea could be right). I need to keep looking and reading to find out more about the dynamic properties of DNA molecules under stress.