














As I wrote a few days ago, our attempts to send pools of transformed Haemophilus influenzae cells to London (to be tested by our collaborators there) were thwarted by FedEx, and we're now considering doing the experiment ourselves.
The experimental plan is to incubate pools of transformed bacteria in human blood and look for survivors. The bacterial cells are of the standard lab strain, which is rapidly killed by factors in blood. The pools consisted of cells that had been transformed with fragments of chromosomal DNA from a strain that survives just fine in blood, and we're hoping that any survivors from our transformed pools will have acquired the gene or genes responsible for survival.
The RA did her PhD in the London lab, and she has done such assays when she was there. BUT big logistic and administrative problems are created by research using blood. The London lab uses heparin-treated blood freshly drawn from a lab volunteer. I'd be happy to volunteer - needles don't bother me, and although I don't have the survival-assay protocol I don't think very much blood is needed. But getting permission to take blood from any of us would require obtaining permission from the university's 'Ethics of Human Experimentation' committee. And none of us knows how to draw blood...
UBC has a Centre for Blood Research, located just upstairs in our building - they study, among other things, ways to improve collection and use of blood and blood products. I wonder if we might be able to get blood from them?
Experiments using human blood almost certainly also have lab-safety issues, and we'd probably need to get a new permit especially for this work. The post-doc thinks the regulations aren't very onerous - we'd just have to work in a biosafety hood, and the group next door has one they don't use very often. But I'd need to check this too.
Another possibility is using commercial human serum rather than blood. When I wrote a post about this phenomenon last month, I showed a graph from an experiment that used human serum, not blood. I just went back and checked their Methods section; that serum had been obtained by the researchers from volunteer blood that was allowed to clot normally, and stored frozen at -70 °C. But normal human serum can be purchased from various suppliers. Hmm, so can normal human blood... I'll have to ask our London collaborators whether they use fresh blood because frozen blood and serum don't work well.
Of course, if we don't send our cells to our London collaborators, they'll have to send their blood-resistant strain to us so we have the right positive control for the experiments. They'd have to deal with the shipping problem, but we'd have to first obtain an importation permit. Luckily we know how to do that, and it normally only takes a few days (once the Christmas disruption is over). I just checked the catalog of the American Type Culture Collection, and we can't get the strain we need from them.
Field of Science
-
-
Change of address1 year ago in Variety of Life
-
Change of address1 year ago in Catalogue of Organisms
-
-
Earth Day: Pogo and our responsibility1 year ago in Doc Madhattan
Writing the recombination tract paper
The post-doc and I are finally writing his paper on his Illumina sequencing analysis of recombinant segments in transformant genomes.
I was initially optimistic that we'd get it done really fast, since he's done so much thinking and analysis and writing about it in other contexts (mainly grant and fellowship applications). But the thinking and analysis and writing are all so complex that we still have to do a lot specifically for the paper.
We're pretty well balanced in our approaches; he's mostly trying to make everything as accurate as possible (for the genomics expert readers), and I'm mostly trying to make everything as transparent as possible (for the non-genomics readers). We're both pretty argumentative, so passers-by often see the fur flying, but we're both reasonable enough that we can usually discover why we disagree and find a solution that we both like.
I was initially optimistic that we'd get it done really fast, since he's done so much thinking and analysis and writing about it in other contexts (mainly grant and fellowship applications). But the thinking and analysis and writing are all so complex that we still have to do a lot specifically for the paper.
We're pretty well balanced in our approaches; he's mostly trying to make everything as accurate as possible (for the genomics expert readers), and I'm mostly trying to make everything as transparent as possible (for the non-genomics readers). We're both pretty argumentative, so passers-by often see the fur flying, but we're both reasonable enough that we can usually discover why we disagree and find a solution that we both like.
FedEx, why oh why do you hate us so?
The cells we're trying to ship to London (post 1, post 2) are back in our freezer.
When I last blogged about them (on Monday) they had been sent back to us by the local FedEx office because the paperwork wasn't perfect and we needed to put the styrofoam container into an appropriately labeled cardboard box. We'd fixed the errors (not all ours) and were only waiting to get some dry ice from Chemistry Stores on Tuesday.
So we sent the cells off on Tuesday with 8 kg of dry ice, and on Wednesday they came back again! This time there were only three tiny errors in the paperwork, only one of which was our oversight. But the errors were easy to fix (at least after I again called the helpful people at FedEx's Dangerous Goods office), so we topped up the dry ice and sent the shipment off again (third time) on Wednesday afternoon.
It didn't come back on Thursday morning (premature sigh of relief), but when we tracked its progress on Friday morning we found that, although it had been accepted by the Vancouver airport office in Richmond and shipped off to their Memphis hub, Memphis had sent it back to Richmond! It had arrived there early on Friday morning and was awaiting clearance (customs?).
I thought it would then be delivered back to us, but when it hadn't shown up by Friday afternoon I had to rush to the airport to fetch it, because I didn't want the cells to thaw out over the weekend. The clerk at the airport FedEx office showed me the tracking record in his system. The package had been rejected by the Memphis Ramp Agent, without any explanation. The clerk couldn't find anything wrong with the paperwork or packaging, except maybe that the 'dangerous goods' status should have been indicated on the computer-generated waybill. He said that it's very tricky to do this annotation on a computer-generated waybill, and kindly gave me a stack of paper/carbon copy waybills to use next time. On Monday I'll call the Dangerous Goods office again, to see if they either agree with this or have another explanation.
To add insult to injury, the RA says that FedEx is charging us the full price (more than $200) for each failed shipment!
Will we try again? Maybe not. The researcher who was going to do the assays in London is about to leave for a post-doc position at Stanford. And the RA says that we should just do the assays here, because she's done them before and they're not difficult.
Monday morning update: I just spoke to a FedEx Dangerous Goods expert. He said that the UK will not accept any shipments containing agents infectious to humans! My London colleagues had assured me that no import permit was needed for Haemophilus influenzae, and this document appears to confirm that, since H. influenzae is in Hazard Group 2.
Another update: I called FedEx back - it's a UK FedEx rule, not a UK government rule. Too bad they didn't bother to tell me this any of the other three times I spoke to them about this shipment; they'd have saved us a lot of money.
When I last blogged about them (on Monday) they had been sent back to us by the local FedEx office because the paperwork wasn't perfect and we needed to put the styrofoam container into an appropriately labeled cardboard box. We'd fixed the errors (not all ours) and were only waiting to get some dry ice from Chemistry Stores on Tuesday.
So we sent the cells off on Tuesday with 8 kg of dry ice, and on Wednesday they came back again! This time there were only three tiny errors in the paperwork, only one of which was our oversight. But the errors were easy to fix (at least after I again called the helpful people at FedEx's Dangerous Goods office), so we topped up the dry ice and sent the shipment off again (third time) on Wednesday afternoon.
It didn't come back on Thursday morning (premature sigh of relief), but when we tracked its progress on Friday morning we found that, although it had been accepted by the Vancouver airport office in Richmond and shipped off to their Memphis hub, Memphis had sent it back to Richmond! It had arrived there early on Friday morning and was awaiting clearance (customs?).
I thought it would then be delivered back to us, but when it hadn't shown up by Friday afternoon I had to rush to the airport to fetch it, because I didn't want the cells to thaw out over the weekend. The clerk at the airport FedEx office showed me the tracking record in his system. The package had been rejected by the Memphis Ramp Agent, without any explanation. The clerk couldn't find anything wrong with the paperwork or packaging, except maybe that the 'dangerous goods' status should have been indicated on the computer-generated waybill. He said that it's very tricky to do this annotation on a computer-generated waybill, and kindly gave me a stack of paper/carbon copy waybills to use next time. On Monday I'll call the Dangerous Goods office again, to see if they either agree with this or have another explanation.
To add insult to injury, the RA says that FedEx is charging us the full price (more than $200) for each failed shipment!
Will we try again? Maybe not. The researcher who was going to do the assays in London is about to leave for a post-doc position at Stanford. And the RA says that we should just do the assays here, because she's done them before and they're not difficult.
Monday morning update: I just spoke to a FedEx Dangerous Goods expert. He said that the UK will not accept any shipments containing agents infectious to humans! My London colleagues had assured me that no import permit was needed for Haemophilus influenzae, and this document appears to confirm that, since H. influenzae is in Hazard Group 2.
Another update: I called FedEx back - it's a UK FedEx rule, not a UK government rule. Too bad they didn't bother to tell me this any of the other three times I spoke to them about this shipment; they'd have saved us a lot of money.
Yet another reason why the Wolfe-Simon conclusion is so improbable
Experimental results need to be evaluated from two perspectives, the quality of the data and the probability of the explanation. I and many others have critiqued the Wolfe-Simon results based on the poor quality of the data, and chemists have critiqued them based on the predicted instability of arsenic bonds in a DNA backbone. But I don't think anyone has spelled out the improbability from an evolutionary perspective. So here goes...
We're told that the authors originally thought they might have cultured a member of the hypothetical 'shadow biosphere'. If such organisms existed they would be descendants of an evolutionary lineage that has been evolving independently of known organisms for billions of years. Known organisms all belong to a single lineage, and the 'shadow biosphere organisms might either have originated independently of this lineage (A in the sketch below) or diverged from it shortly after its origin (B in the sketch below).
 The organisms contained DNA, and PCR amplification using 'universal' primers for the 16S rRNA gene was successful. This meant that the organisms could not be members of a type A shadow biosphere, as an independent origin wouldn't have produced either DNA or 16S rRNA genes. But placing the 16S rRNA sequence in a phylogenetic tree showed that the organisms weren't members of a type B shadow biosphere either. Instead they were members of a conventional group of bacteria, indicated by the dark wedge in the bacterial triangle.
The organisms contained DNA, and PCR amplification using 'universal' primers for the 16S rRNA gene was successful. This meant that the organisms could not be members of a type A shadow biosphere, as an independent origin wouldn't have produced either DNA or 16S rRNA genes. But placing the 16S rRNA sequence in a phylogenetic tree showed that the organisms weren't members of a type B shadow biosphere either. Instead they were members of a conventional group of bacteria, indicated by the dark wedge in the bacterial triangle.
Finding that a shadow biosphere organism had DNA with a backbone containing only arsenic (no phosphorus) would have been very surprising, both because we have no evidence that the shadow biosphere exists and because of the predicted instability of the arsenic ester bonds. However, the claim that a conventional bacterium can function when its DNA backbone contains a mixture of arsenic and phosphorus is even more improbable. Here's why:
Many different enzymes interact with DNA, and almost all of them need to do so with high precision. They can do this because the DNA backbone has a very consistent structure. Although the four bases that attach to the backbone have different shapes, they can occur with any sequence only because the nature of the base pair does not alter the backbone geometry. The enzyme DNA polymerase is especially dependent on precise interactions with DNA, because it is responsible for accurate replication of the genetic material. Typical measures of the fidelity of DNA replication by bacterial DNA polymerases show an error rate of about 10^-9.
The covalent radius of arsenic is about 11% larger than that of phosphorus (see this periodic table tool), and the As-O bond lengths reported in Table 3 are about 13% longer than the P-O bond lengths in DNA (Table S3). No DNA polymerase could function on a template with such unpredictable geometry. Even if it could bind to the template strand and proceed along it, the differing bond lengths it encountered would prevent the incoming nucleotides from forming the necessary bond with the growing end. And even if it could create the backbone of the daughter strand, the error rate due to base mispairing would be so high that reproduction would fail.
Similar problems would affect every enzyme that contributes to production of the DNA and RNA precursors, to DNA and RNA synthesis, to DNA repair, and to energy metabolism. No matter how stable the arsenic-substituted metabolites might be (and the chemists tell us they are mostly very unstable), the proteins that need to interact with them would not be able to cope with the unpredictability of usinging both arsenic and phosphorus versions.
The fundamental problem isn't that millions of generations would be needed to evolve enzymes that could tolerate a mixture of arsenic and phosphorus, but that the required adaptations are not compatible with the precision needed for function.
We're told that the authors originally thought they might have cultured a member of the hypothetical 'shadow biosphere'. If such organisms existed they would be descendants of an evolutionary lineage that has been evolving independently of known organisms for billions of years. Known organisms all belong to a single lineage, and the 'shadow biosphere organisms might either have originated independently of this lineage (A in the sketch below) or diverged from it shortly after its origin (B in the sketch below).

Finding that a shadow biosphere organism had DNA with a backbone containing only arsenic (no phosphorus) would have been very surprising, both because we have no evidence that the shadow biosphere exists and because of the predicted instability of the arsenic ester bonds. However, the claim that a conventional bacterium can function when its DNA backbone contains a mixture of arsenic and phosphorus is even more improbable. Here's why:
Many different enzymes interact with DNA, and almost all of them need to do so with high precision. They can do this because the DNA backbone has a very consistent structure. Although the four bases that attach to the backbone have different shapes, they can occur with any sequence only because the nature of the base pair does not alter the backbone geometry. The enzyme DNA polymerase is especially dependent on precise interactions with DNA, because it is responsible for accurate replication of the genetic material. Typical measures of the fidelity of DNA replication by bacterial DNA polymerases show an error rate of about 10^-9.
The covalent radius of arsenic is about 11% larger than that of phosphorus (see this periodic table tool), and the As-O bond lengths reported in Table 3 are about 13% longer than the P-O bond lengths in DNA (Table S3). No DNA polymerase could function on a template with such unpredictable geometry. Even if it could bind to the template strand and proceed along it, the differing bond lengths it encountered would prevent the incoming nucleotides from forming the necessary bond with the growing end. And even if it could create the backbone of the daughter strand, the error rate due to base mispairing would be so high that reproduction would fail.
Similar problems would affect every enzyme that contributes to production of the DNA and RNA precursors, to DNA and RNA synthesis, to DNA repair, and to energy metabolism. No matter how stable the arsenic-substituted metabolites might be (and the chemists tell us they are mostly very unstable), the proteins that need to interact with them would not be able to cope with the unpredictability of usinging both arsenic and phosphorus versions.
The fundamental problem isn't that millions of generations would be needed to evolve enzymes that could tolerate a mixture of arsenic and phosphorus, but that the required adaptations are not compatible with the precision needed for function.
Comments on Dr. Wolfe-Simon's Response
Preliminary Response to Questions Concerning the Science Article (from F. Wolfe-Simon, Dec. 16, 2010)
(In this post I'm only addressing the specific points made in this Q&A. Magenta numbers identify points I have concerns about. As always, I'm not asking readers to take my word for anything; if you have doubts you should look up the information and check my calculations for yourself.)
Question:
Some people have questioned whether the DNA was sufficiently cleaned by your technique using gel electrophoresis, to separate it from other molecules. Do you feel this is a valid concern?
Answer:
Our DNA extraction and purification protocol begins with washed cells, pelleted from media. These are then subjected to a standard DNA extraction protocol (1), which included multiple phenol chloroform steps to remove impurities, including any unincorporated arsenate (As) (2). After this, the DNA was electrophoresed, further separating the DNA from impurities (3, 4). Any residual As from the media would have been removed by washing the cells prior to extraction (5) and by partitioning into the aqueous phase (6) during the 3 phenol:chloroform steps in the extraction. If As was incorporated into a lipid or protein it would have partitioned into the phenol, phenol:chloroform, or chloroform fractions (7). Additionally, DNA extracted in this manner on other samples was also successfully used in further analyses, including PCR (8), that require highly purified DNA (9).
The arsenic measured by NanoSIMS in the gel band is consistent with our other measurements and another line of evidence.
Our radiolabeled 73AsO43- experiment showed that of the total radiolabel associated with the cell pellet 11.0 % ± 0.1 % was associated with the DNA/RNA fraction (10). This indicated that we should expect some arsenate of the total pool associated with the nucleic acids (11). To interpret these data, we coupled our interpretation with our EXAFS evidence suggesting that intracellular arsenic was As(V) bound to C (12), and was not free in solution as an ion. This suggests the As is in, an organic molecule with bond distances consistent with a chemical environment analogous to phosphate (Figs. 3A, S3 "bond lengths" table). Further supporting our interpretation of the previous mentioned two analyses, we used a third line of evidence from NanoSIMS, a completely different technique from the other two. We find elemental arsenic (as measured by NanoSIMS) associated with the gel band that is more than two times the background in the gel (13). Based on the above discussion, we do not feel this is a valid concern.
My concerns:
(1) The DNA extraction procedure included only some of the components of a standard DNA extraction protocol. First, only a single ethanol precipitation was done, whereas getting relatively pure DNA requires at least two rounds of extraction and precipitation. Second, the pellets were not washed, so that unincorporated arsenate (or phosphate) present in the aqueous fractions may have been precipitated with the DNA, and may also have been present in the alcohol supernatants contaminating the pellets. Third, no column clean-up step was done.
(2) This statement implies that unincorporated arsenate in the cell lysate would partition into the phenol and chloroform. This seems a priori improbable, as arsenate is very soluble in water. No controls were done to find out how unincorporated arsenate or phosphate would partition in these extractions.
(3) Gel electrophoresis can remove impurities but it is not guaranteed to do so. Any impurities that migrate at a similar rate to the DNA, or are electrostatically associated with it, will be present in the gel slice. Any impurities that diffuse into the gel buffer may become distributed throughout the gel. Any impurities already present present in the agarose or gel buffer will also be in the gel slices. These concerns are strengthened by the failure to purify the DNA away from the gel slice (see (13) below).
(4) No control was performed for non-covalent association of arsenate (or phosphate) with DNA. In another control extraction, arsenate and lysis solution should have been mixed with previously purified DNA from E. coli or other phosphate-grown cells, to see if any arsenic co-purified with the DNA.
(5) No control was performed for the effectiveness of this washing. E. coli or other cells grown in the absence of arsenate should have been mixed with the +As/-P medium and then subject to the same washing and extraction steps.
(6) Here arsenate is predicted to partition into the aqueous phase. Is this the same arsenate that partitioned into the phenol and chloroform in (2) above?
(7) In the ICP-MS analysis presented in Table S1, almost all the arsenic did partition into the phenol phase, and almost as much arsenic was present in the phenol fraction of the phosphate-grown cell (4725 vs 3683 ppb). In fact, the aqueous phase of arsenate-grown cells contained no detectable arsenic at all, even though this is the fraction from which the arsenic-containing DNA was precipitated.
(8) The online Methods say that DNA from all growth conditions, worked fine in the PCR reactions used for the phylogenetic analysis. This strongly suggests that the DNA from arsenate-grown cells has a normal phosphorus backbone. The polymerases used for PCR have very high fidelity and would not tolerate substitution of arsenic for phosphorus.
(9) PCR does not require purified DNA; it even works very well on whole-cell lysates.
(10) See point (7)
(11) But this pool is expected to contain all of the water-soluble constituents of the cell. The elemental analysis report that the arsenic was bonded to carbon doesn't mean that it is bonded to DNA. And most of this arsenic partitioned into the phenol phase - is it thought to be lipid? If so, maybe the arsenic is bonded to C in lipids.
(12) Wait! IANAC (I am not a chemist), but if the 'intracellular arsenic was As(V) bound to C' then it couldn't be arsenic incorporated into DNA or RNA, as it would then have to be bound to O in DNA's diester backbone.
(13) Let's think more about the arsenic in the gel bands. The whole gel slices were assayed (the DNA was not purified away from the agarose); since the gel is 1% agarose and a gel slice is unlikely to weigh less than 100 mg, each slice would contain at least 1 mg of agarose. The DNA bands in Fig. 2A are unlikely to contain more than 1 µg of DNA, probably less for arsenate-grown DNA in lane 2). Thus we can generously assume that 99.9% of the carbon in each DNA sample came from the agarose, and no more than 0.1% from the DNA. According to the figure legend and the numbers at the bottoms of the gel lanes the arsenate-grown sample had 13.4 atoms of arsenic per 10^6 atoms of carbon. This is 13.4 arsenic atoms per 1000 DNA carbons. Since A, G and T nucleotides contains 10 carbons and C has 9, this is 13.4 arsenic atoms per 102.6 nucleotides, or about 26 per 100 base pairs. That's quite a lot of arsenic. Even more surprising, the phosphate-grown sample had 6.9 arsenics per 10^6 carbons, which would be about 14 arsenics per 100 bp. The gel blank had even more arsenic, and three times as much phosphorus. This strongly suggests that the gel was contaminated with both arsenic and phosphorus, perhaps introduced with the DNA samples. Until such contamination can be ruled out, the two-fold higher arsenic concentration and three-fold lower phosphorus concentration associated with the arsenic-grown DNA sample cannot be seen as significant.
Question:
Others have argued that arsenate-linked DNA should have quickly fallen apart when exposed to water. Could you address this?
Answer:
We are not aware of any studies that address arsenate bound in long chain polyesters or nucleotide di- or tri-esters of arsenate, which would be directly relevant to our study. Published studies have shown that simple arsenic esters have much higher hydrolysis rates than phosphate esters (1-3). The experiments published to date have specifically looked at the exchange or hydrolysis of alkyl tri-esters of arsenate [Eqn. 1] and alkyl di-esters of arsenite [Eqn. 2]:
OAs(OR)3 + H2O → OAs(OH)(OR)2+ ROH [1]
OAs(OH)(OR)2 + H2O → OAs(OH)2(OR) + ROH [2]
where R = methyl, ethyl, n-pentyl and isopropyl. Reference 2 demonstrated that the hydrolysis rates for these simple alkyl triesters of arsenate decreased with increasing carbon chain length (complexity) of the alkyl substituent (methyl > ethyl > n-pentyl > isopropyl) (14). No work has been done on the hydrolysis rates of arsenate-linked nucleotides or other biologically relevant moieties.
If the hydrolytic rate trend reported in Ref. 2 continues to larger-weight organics, such as those found in biomolecules, it is conceivable that arsenate-linked biopolymers might be more resistant to hydrolysis than previously thought (15). The small model compounds investigated in Refs. 1-3 are relatively flexible and can easily adopt the ideal geometry for water to attack the arseno-ester bond (16). Arsenate esters of large, bio-molecules, however, are likely to be more sterically hindered leading to slower rates of hydrolysis (17).
This type of steric constraint on reaction rate accounts for the wide range of rates seen in the behavior of some phosphate linked nucleotides. In small ribozymes, the phophodiester linkages at the site of catalysis can be hydrolyzed on the order of tens of seconds (with a chemical rate of 1 s-1). This rate enhancement is achieved by orienting the linkage for in-line attack by a nucleophile (an adjacent 2' hydroxyl group). Moreover, the autodegradation patterns are consistent with specific base composition. On the other hand, the hydrolysis rates for phosphodiester bonds in A form duplexes of RNA are many orders of magnitude slower, because these linkages cannot access easily the geometry necessary for hydrolysis.
The rates in DNA may be much slower than model compounds because of the geometrical constraints imposed upon the backbone by the helix (18).
The kinetics of the hydrolysis of arsenate-linked biopolymers is clearly an area where more research is warranted.
My concerns:
(14) Again IANAC. But note that these are hydrocarbons and thus quite hydrophobic, especially the pentyl chain (5 carbons) and the isopropyl chain (3 branching carbons). Attaching three of these by ester bonds to the arsenate effectively surrounds the bonds with hydrophobic shells that exclude water. Thus it's not surprising that the hydrolysis reaction occurs less often.
(15) But if the increased stability described in Reference 2 (yes, I looked at this paper, and I'm trying to get hold of the 1870 reference too) is due to increasing hydrophobicity of the ester bonds' environment, then the effect will not extrapolate to long hydrophilic biological molecules such as DNA.
(16) Not if they're surrounded by a hydrophobic shell, and probably also associating with each other to reduce the shell's exposure to the aqueous solvent.
(17) DNA has its backbone on the outside of the double helix, and the entire molecule is quite hydrophilic.
(18) Assuming the overall structure of arsenic bonds in DNA is like that of phosphorus bonds in DNA, shouldn't any stability-enhancing geometrical constraints of the DNA structure be experienced by both phosphorus and arsenic bonds? We would then still expect arsenic bonds in DNA to be 100-fold less stable than phosphate bonds.
Question:
Is it possible that salts in your growth media could have provided enough trace phosphorus to sustain the bacteria?
Answer:
The data and sample labeling in Table S1 has caused some confusion. To clarify, for every experiment, a single batch of artificial Mono Lake water was made with the following formulation: AML60 salts, no P, no As, no glucose, no vitamins. Table S1 shows examples of ICPMS measurements of elemental phosphorus (~3 µM) and arsenate made on this formulation prior to any further additions (19, 20). Then we added glucose and vitamins for all three treatments and either As for the +As treatments or P for the +P treatments. The P measurements made on the medium after the addition of sucrose and vitamins and after addition of As were also ~3 µM in this batch. Therefore, it was clear that any P impurity that was measured (~3 µM, this was the high range) came in with the major salts, and that all experiments contain identical P background (including any P brought in with the culture inocula).
In the Science paper, we show data from one experiment of many replicated experiments that demonstrates no growth of cells in media without added arsenate or phosphate (Figure 1). These data clearly demonstrate that strain GFAJ-1 was unable to utilize the 3µM P to support further growth in the absence of arsenate (21). Moreover, the intracellular P content determined for the +As/-P grown cells was not enough to support the full requirement of P for cellular function (22).
Note on culturing: All experiments were initiated with inocula from sustained +As/-P conditions. Prior to the experiments, the cells had been grown long term, for multiple generations from a single colony grown on solid media with no added phosphate. Before this, they were grown as an enrichment for more than 10 transfers and always into new medium that was +As/-P. We therefore feel that there is not significant carry-over of P. We also argue that there would not have been enough cellular P to support additional growth based on an internal recycling pool of P (23).
My concerns:
(19) The two batches of AML60 salts assayed contained 3.7 and <0.3 µM P, and the single batch of cell wash solution contained 7.4 µM P. Given this variability, the similarity of the two batches of -P/+As medium doesn't inspire much confidence.
(20) Was no effort made to identify and eliminate the source(s) of this contamination?
(21) Agreed, with the proviso that the media be tested and shown to be identical except for the phosphate and arsenate. But this wouldn't mean that arsenic replaced phosphorus in any biological molecules in GFAJ cells, just that the cells needed arsenate for something.
(22) Did this calculation take into account the very high carbon content of the poly-hydroxybutyrate granules in these cells? PHB can account for up to 90% of the dry weight of phosphate-starved cells, and its carbon will skew estimates of C:P ratios.
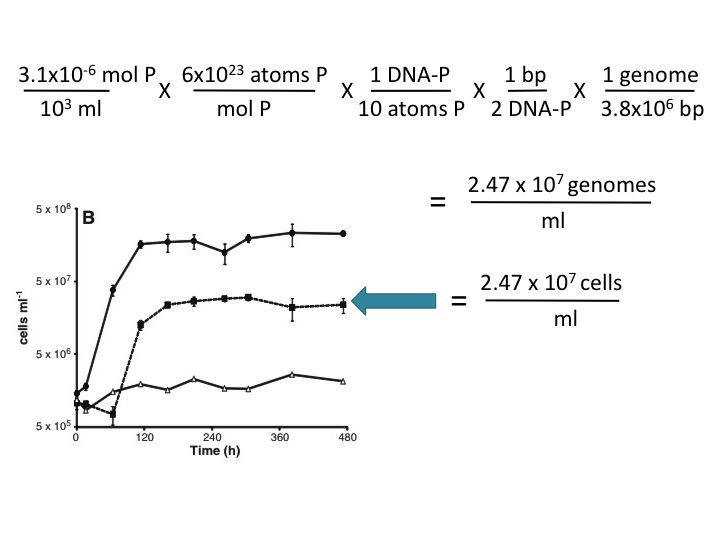
(23) This assertion is not supported here by any evidence, and it is contradicted by the Makino et al. (2003) reference cited by the Science paper (ref. 13). These authors found that 10% of the P in phosphate-limited E. coli is in the DNA and the rest is in RNA and other cellular components. Using this value and your own estimate of genome size (3.8 Mbp), ~3 µM P is sufficient to account for the observed growth in the -P/+As medium. Here's the calculation:

(In this post I'm only addressing the specific points made in this Q&A. Magenta numbers identify points I have concerns about. As always, I'm not asking readers to take my word for anything; if you have doubts you should look up the information and check my calculations for yourself.)
Question:
Some people have questioned whether the DNA was sufficiently cleaned by your technique using gel electrophoresis, to separate it from other molecules. Do you feel this is a valid concern?
Answer:
Our DNA extraction and purification protocol begins with washed cells, pelleted from media. These are then subjected to a standard DNA extraction protocol (1), which included multiple phenol chloroform steps to remove impurities, including any unincorporated arsenate (As) (2). After this, the DNA was electrophoresed, further separating the DNA from impurities (3, 4). Any residual As from the media would have been removed by washing the cells prior to extraction (5) and by partitioning into the aqueous phase (6) during the 3 phenol:chloroform steps in the extraction. If As was incorporated into a lipid or protein it would have partitioned into the phenol, phenol:chloroform, or chloroform fractions (7). Additionally, DNA extracted in this manner on other samples was also successfully used in further analyses, including PCR (8), that require highly purified DNA (9).
The arsenic measured by NanoSIMS in the gel band is consistent with our other measurements and another line of evidence.
Our radiolabeled 73AsO43- experiment showed that of the total radiolabel associated with the cell pellet 11.0 % ± 0.1 % was associated with the DNA/RNA fraction (10). This indicated that we should expect some arsenate of the total pool associated with the nucleic acids (11). To interpret these data, we coupled our interpretation with our EXAFS evidence suggesting that intracellular arsenic was As(V) bound to C (12), and was not free in solution as an ion. This suggests the As is in, an organic molecule with bond distances consistent with a chemical environment analogous to phosphate (Figs. 3A, S3 "bond lengths" table). Further supporting our interpretation of the previous mentioned two analyses, we used a third line of evidence from NanoSIMS, a completely different technique from the other two. We find elemental arsenic (as measured by NanoSIMS) associated with the gel band that is more than two times the background in the gel (13). Based on the above discussion, we do not feel this is a valid concern.
My concerns:
(1) The DNA extraction procedure included only some of the components of a standard DNA extraction protocol. First, only a single ethanol precipitation was done, whereas getting relatively pure DNA requires at least two rounds of extraction and precipitation. Second, the pellets were not washed, so that unincorporated arsenate (or phosphate) present in the aqueous fractions may have been precipitated with the DNA, and may also have been present in the alcohol supernatants contaminating the pellets. Third, no column clean-up step was done.
(2) This statement implies that unincorporated arsenate in the cell lysate would partition into the phenol and chloroform. This seems a priori improbable, as arsenate is very soluble in water. No controls were done to find out how unincorporated arsenate or phosphate would partition in these extractions.
(3) Gel electrophoresis can remove impurities but it is not guaranteed to do so. Any impurities that migrate at a similar rate to the DNA, or are electrostatically associated with it, will be present in the gel slice. Any impurities that diffuse into the gel buffer may become distributed throughout the gel. Any impurities already present present in the agarose or gel buffer will also be in the gel slices. These concerns are strengthened by the failure to purify the DNA away from the gel slice (see (13) below).
(4) No control was performed for non-covalent association of arsenate (or phosphate) with DNA. In another control extraction, arsenate and lysis solution should have been mixed with previously purified DNA from E. coli or other phosphate-grown cells, to see if any arsenic co-purified with the DNA.
(5) No control was performed for the effectiveness of this washing. E. coli or other cells grown in the absence of arsenate should have been mixed with the +As/-P medium and then subject to the same washing and extraction steps.
(6) Here arsenate is predicted to partition into the aqueous phase. Is this the same arsenate that partitioned into the phenol and chloroform in (2) above?
(7) In the ICP-MS analysis presented in Table S1, almost all the arsenic did partition into the phenol phase, and almost as much arsenic was present in the phenol fraction of the phosphate-grown cell (4725 vs 3683 ppb). In fact, the aqueous phase of arsenate-grown cells contained no detectable arsenic at all, even though this is the fraction from which the arsenic-containing DNA was precipitated.
(8) The online Methods say that DNA from all growth conditions, worked fine in the PCR reactions used for the phylogenetic analysis. This strongly suggests that the DNA from arsenate-grown cells has a normal phosphorus backbone. The polymerases used for PCR have very high fidelity and would not tolerate substitution of arsenic for phosphorus.
(9) PCR does not require purified DNA; it even works very well on whole-cell lysates.
(10) See point (7)
(11) But this pool is expected to contain all of the water-soluble constituents of the cell. The elemental analysis report that the arsenic was bonded to carbon doesn't mean that it is bonded to DNA. And most of this arsenic partitioned into the phenol phase - is it thought to be lipid? If so, maybe the arsenic is bonded to C in lipids.
(12) Wait! IANAC (I am not a chemist), but if the 'intracellular arsenic was As(V) bound to C' then it couldn't be arsenic incorporated into DNA or RNA, as it would then have to be bound to O in DNA's diester backbone.
(13) Let's think more about the arsenic in the gel bands. The whole gel slices were assayed (the DNA was not purified away from the agarose); since the gel is 1% agarose and a gel slice is unlikely to weigh less than 100 mg, each slice would contain at least 1 mg of agarose. The DNA bands in Fig. 2A are unlikely to contain more than 1 µg of DNA, probably less for arsenate-grown DNA in lane 2). Thus we can generously assume that 99.9% of the carbon in each DNA sample came from the agarose, and no more than 0.1% from the DNA. According to the figure legend and the numbers at the bottoms of the gel lanes the arsenate-grown sample had 13.4 atoms of arsenic per 10^6 atoms of carbon. This is 13.4 arsenic atoms per 1000 DNA carbons. Since A, G and T nucleotides contains 10 carbons and C has 9, this is 13.4 arsenic atoms per 102.6 nucleotides, or about 26 per 100 base pairs. That's quite a lot of arsenic. Even more surprising, the phosphate-grown sample had 6.9 arsenics per 10^6 carbons, which would be about 14 arsenics per 100 bp. The gel blank had even more arsenic, and three times as much phosphorus. This strongly suggests that the gel was contaminated with both arsenic and phosphorus, perhaps introduced with the DNA samples. Until such contamination can be ruled out, the two-fold higher arsenic concentration and three-fold lower phosphorus concentration associated with the arsenic-grown DNA sample cannot be seen as significant.
Question:
Others have argued that arsenate-linked DNA should have quickly fallen apart when exposed to water. Could you address this?
Answer:
We are not aware of any studies that address arsenate bound in long chain polyesters or nucleotide di- or tri-esters of arsenate, which would be directly relevant to our study. Published studies have shown that simple arsenic esters have much higher hydrolysis rates than phosphate esters (1-3). The experiments published to date have specifically looked at the exchange or hydrolysis of alkyl tri-esters of arsenate [Eqn. 1] and alkyl di-esters of arsenite [Eqn. 2]:
OAs(OR)3 + H2O → OAs(OH)(OR)2+ ROH [1]
OAs(OH)(OR)2 + H2O → OAs(OH)2(OR) + ROH [2]
where R = methyl, ethyl, n-pentyl and isopropyl. Reference 2 demonstrated that the hydrolysis rates for these simple alkyl triesters of arsenate decreased with increasing carbon chain length (complexity) of the alkyl substituent (methyl > ethyl > n-pentyl > isopropyl) (14). No work has been done on the hydrolysis rates of arsenate-linked nucleotides or other biologically relevant moieties.
If the hydrolytic rate trend reported in Ref. 2 continues to larger-weight organics, such as those found in biomolecules, it is conceivable that arsenate-linked biopolymers might be more resistant to hydrolysis than previously thought (15). The small model compounds investigated in Refs. 1-3 are relatively flexible and can easily adopt the ideal geometry for water to attack the arseno-ester bond (16). Arsenate esters of large, bio-molecules, however, are likely to be more sterically hindered leading to slower rates of hydrolysis (17).
This type of steric constraint on reaction rate accounts for the wide range of rates seen in the behavior of some phosphate linked nucleotides. In small ribozymes, the phophodiester linkages at the site of catalysis can be hydrolyzed on the order of tens of seconds (with a chemical rate of 1 s-1). This rate enhancement is achieved by orienting the linkage for in-line attack by a nucleophile (an adjacent 2' hydroxyl group). Moreover, the autodegradation patterns are consistent with specific base composition. On the other hand, the hydrolysis rates for phosphodiester bonds in A form duplexes of RNA are many orders of magnitude slower, because these linkages cannot access easily the geometry necessary for hydrolysis.
The rates in DNA may be much slower than model compounds because of the geometrical constraints imposed upon the backbone by the helix (18).
The kinetics of the hydrolysis of arsenate-linked biopolymers is clearly an area where more research is warranted.
My concerns:
(14) Again IANAC. But note that these are hydrocarbons and thus quite hydrophobic, especially the pentyl chain (5 carbons) and the isopropyl chain (3 branching carbons). Attaching three of these by ester bonds to the arsenate effectively surrounds the bonds with hydrophobic shells that exclude water. Thus it's not surprising that the hydrolysis reaction occurs less often.
(15) But if the increased stability described in Reference 2 (yes, I looked at this paper, and I'm trying to get hold of the 1870 reference too) is due to increasing hydrophobicity of the ester bonds' environment, then the effect will not extrapolate to long hydrophilic biological molecules such as DNA.
(16) Not if they're surrounded by a hydrophobic shell, and probably also associating with each other to reduce the shell's exposure to the aqueous solvent.
(17) DNA has its backbone on the outside of the double helix, and the entire molecule is quite hydrophilic.
(18) Assuming the overall structure of arsenic bonds in DNA is like that of phosphorus bonds in DNA, shouldn't any stability-enhancing geometrical constraints of the DNA structure be experienced by both phosphorus and arsenic bonds? We would then still expect arsenic bonds in DNA to be 100-fold less stable than phosphate bonds.
Question:
Is it possible that salts in your growth media could have provided enough trace phosphorus to sustain the bacteria?
Answer:
The data and sample labeling in Table S1 has caused some confusion. To clarify, for every experiment, a single batch of artificial Mono Lake water was made with the following formulation: AML60 salts, no P, no As, no glucose, no vitamins. Table S1 shows examples of ICPMS measurements of elemental phosphorus (~3 µM) and arsenate made on this formulation prior to any further additions (19, 20). Then we added glucose and vitamins for all three treatments and either As for the +As treatments or P for the +P treatments. The P measurements made on the medium after the addition of sucrose and vitamins and after addition of As were also ~3 µM in this batch. Therefore, it was clear that any P impurity that was measured (~3 µM, this was the high range) came in with the major salts, and that all experiments contain identical P background (including any P brought in with the culture inocula).
In the Science paper, we show data from one experiment of many replicated experiments that demonstrates no growth of cells in media without added arsenate or phosphate (Figure 1). These data clearly demonstrate that strain GFAJ-1 was unable to utilize the 3µM P to support further growth in the absence of arsenate (21). Moreover, the intracellular P content determined for the +As/-P grown cells was not enough to support the full requirement of P for cellular function (22).
Note on culturing: All experiments were initiated with inocula from sustained +As/-P conditions. Prior to the experiments, the cells had been grown long term, for multiple generations from a single colony grown on solid media with no added phosphate. Before this, they were grown as an enrichment for more than 10 transfers and always into new medium that was +As/-P. We therefore feel that there is not significant carry-over of P. We also argue that there would not have been enough cellular P to support additional growth based on an internal recycling pool of P (23).
My concerns:
(19) The two batches of AML60 salts assayed contained 3.7 and <0.3 µM P, and the single batch of cell wash solution contained 7.4 µM P. Given this variability, the similarity of the two batches of -P/+As medium doesn't inspire much confidence.
(20) Was no effort made to identify and eliminate the source(s) of this contamination?
(21) Agreed, with the proviso that the media be tested and shown to be identical except for the phosphate and arsenate. But this wouldn't mean that arsenic replaced phosphorus in any biological molecules in GFAJ cells, just that the cells needed arsenate for something.
(22) Did this calculation take into account the very high carbon content of the poly-hydroxybutyrate granules in these cells? PHB can account for up to 90% of the dry weight of phosphate-starved cells, and its carbon will skew estimates of C:P ratios.
(23) This assertion is not supported here by any evidence, and it is contradicted by the Makino et al. (2003) reference cited by the Science paper (ref. 13). These authors found that 10% of the P in phosphate-limited E. coli is in the DNA and the rest is in RNA and other cellular components. Using this value and your own estimate of genome size (3.8 Mbp), ~3 µM P is sufficient to account for the observed growth in the -P/+As medium. Here's the calculation:
The FedEx saga continues
Regular readers (all 5 of you) may remember last week's unsuccessful attempt to ship some of our bacteria to London. FedEx returned our package to us because we hadn't met all their stringent requirements for infections bacteria.
This afternoon we went back to Shipping and Receiving with all the new correct FedEx paperwork and our 24 vials of bacterial cells. The vials were in a plastic box, in a special leakproof container, in this container's specially labeled cardboard carton, in a big styrofoam container for the dry ice, in a very big cardboard box labeled OVERPACK.
Only to learn that Shipping and Receiving had run out of dry ice!
It was too late to rush over to Chemistry Stores to get dry ice, as we'd miss the FedEx pickup time. I briefly considered finding an open-late FedEx office and hand-delivering the package to them. This would have involved sending someone over to Chemistry Stores to get the dry ice while I rode my bike home (5 miles) to get my car. But then I remembered that I'm probably not authorized to even touch this hazardous-goods shipment, much less drive it around town.
Shipping and Receiving won't have dry ice until the day after tomorrow, so tomorrow one of us will go over to Chemistry Stores and get the dry ice we need, and we'll try yet again to get the shipment on its way to London.
This afternoon we went back to Shipping and Receiving with all the new correct FedEx paperwork and our 24 vials of bacterial cells. The vials were in a plastic box, in a special leakproof container, in this container's specially labeled cardboard carton, in a big styrofoam container for the dry ice, in a very big cardboard box labeled OVERPACK.
Only to learn that Shipping and Receiving had run out of dry ice!
It was too late to rush over to Chemistry Stores to get dry ice, as we'd miss the FedEx pickup time. I briefly considered finding an open-late FedEx office and hand-delivering the package to them. This would have involved sending someone over to Chemistry Stores to get the dry ice while I rode my bike home (5 miles) to get my car. But then I remembered that I'm probably not authorized to even touch this hazardous-goods shipment, much less drive it around town.
Shipping and Receiving won't have dry ice until the day after tomorrow, so tomorrow one of us will go over to Chemistry Stores and get the dry ice we need, and we'll try yet again to get the shipment on its way to London.
How to harness distributed discussion of research papers
In this post I'm going to elaborate on a suggestion I saw a few days ago, in an article discussing the role of post-publication commentary in science. (And yes, I'm searching for the source of this idea - if any reader remembers whose it was, please point me to it.)
In modern but pre-internet days, researchers did the research, wrote the paper, submitted it to peer review, made changes, and published it. Other researchers them evaluated this information, using to guide their own work, and discussed its strengths and weaknesses when they cited it in their own papers.
Published papers were also discussed less formally with colleagues, both before and after publication, face-to-face and by mail and phone, and in journal-club presentations and seminars. The ideas from these discussions were incorporated into the formal papers drawing on this work, but they weren't available to anyone but the direct participants.
Now that we're all on line, published papers are also being discussed more publicly, in blogs and other places. Such discussions are extraordinarily valuable for the progress of science - they're written public evaluations, drawn from a wide range of expertise, and usually greatly enriched by comments from and links other researchers. But these pages are all over the place, and finding them requires a lot of active searching.
The Research Blogging site is helping with this problem, by aggregating blog posts that discuss individual research papers. But they can only link to posts that actively insert their code, and so miss quite a lot of the public commentary. So far the journals don't link their papers to this site, so readers who go looking for the paper don't usually think to also check Research Blogging.
A few forward-thinking online journals (PLoS and BMC groups, I'm talking about you) provide their own Comments thread for each paper, so other researchers can provide informal but public feedback . But the researchers don't use these, saying that they don't feel comfortable doing this publicly, or that they don't like the bother of having to register and log on. I know that's true for me, thought I don't know why - I'll happily blog about a paper I've read, but I almost never post comments on its official Comments page.
Sites like The Third Reviewer have tried to solve this by providing journal-independent sites where researchers can post comments about papers. But we won't use these either -the massive wave of discussion about the Wolfe-Simon paper on arsenic bacteria let to exactly zero comments on The Third Reviewer.
Most journals already provide, with each paper they've published, a list of links to the more recent papers that cite it. The suggestion I really liked was that the journals should also aggregate the informal commentary, by providing a separate list of links to ALL the web pages that have link to the paper. Journals could then stop fighting our unwillingness to post comments centrally, and just use our distributed posts to add value to the papers they publish.
I don't think this would be very difficult; lots of sites already have a 'Who links here?' feature, and I think both Google and Yahoo searches can be restricted to sites containing a specific url. The journals could include an explicit disclaimer that the journal in no way vouches for the value or creditability of the information in these links. A few bloggers might have to become a bit more circumspect (probably not a bad thing), and any blogger who didn't want their post linked to the paper could provide a citation to the paper but not link to it.
Jon Eisen, do you think this could work? If PLoS leads, I bet the others will follow.
In modern but pre-internet days, researchers did the research, wrote the paper, submitted it to peer review, made changes, and published it. Other researchers them evaluated this information, using to guide their own work, and discussed its strengths and weaknesses when they cited it in their own papers.
Published papers were also discussed less formally with colleagues, both before and after publication, face-to-face and by mail and phone, and in journal-club presentations and seminars. The ideas from these discussions were incorporated into the formal papers drawing on this work, but they weren't available to anyone but the direct participants.
Now that we're all on line, published papers are also being discussed more publicly, in blogs and other places. Such discussions are extraordinarily valuable for the progress of science - they're written public evaluations, drawn from a wide range of expertise, and usually greatly enriched by comments from and links other researchers. But these pages are all over the place, and finding them requires a lot of active searching.
The Research Blogging site is helping with this problem, by aggregating blog posts that discuss individual research papers. But they can only link to posts that actively insert their code, and so miss quite a lot of the public commentary. So far the journals don't link their papers to this site, so readers who go looking for the paper don't usually think to also check Research Blogging.
A few forward-thinking online journals (PLoS and BMC groups, I'm talking about you) provide their own Comments thread for each paper, so other researchers can provide informal but public feedback . But the researchers don't use these, saying that they don't feel comfortable doing this publicly, or that they don't like the bother of having to register and log on. I know that's true for me, thought I don't know why - I'll happily blog about a paper I've read, but I almost never post comments on its official Comments page.
Sites like The Third Reviewer have tried to solve this by providing journal-independent sites where researchers can post comments about papers. But we won't use these either -the massive wave of discussion about the Wolfe-Simon paper on arsenic bacteria let to exactly zero comments on The Third Reviewer.
Most journals already provide, with each paper they've published, a list of links to the more recent papers that cite it. The suggestion I really liked was that the journals should also aggregate the informal commentary, by providing a separate list of links to ALL the web pages that have link to the paper. Journals could then stop fighting our unwillingness to post comments centrally, and just use our distributed posts to add value to the papers they publish.
I don't think this would be very difficult; lots of sites already have a 'Who links here?' feature, and I think both Google and Yahoo searches can be restricted to sites containing a specific url. The journals could include an explicit disclaimer that the journal in no way vouches for the value or creditability of the information in these links. A few bloggers might have to become a bit more circumspect (probably not a bad thing), and any blogger who didn't want their post linked to the paper could provide a citation to the paper but not link to it.
Jon Eisen, do you think this could work? If PLoS leads, I bet the others will follow.
An apologetic email
Note added Dec. 13: The author of the ABC article has modified it (even though he had accurately quoted what I'd said to him), and he now also quotes from this email.
Dear Dr. Wolfe-Simon,
I'm emailing you to apologize for quotes from me that have just appeared on the ABC News website. I wasn't misquoted, but some of the things I said in a phone interview yesterday morning came across more harshly than I had intended.
I told the interviewer that, even though I think your conclusions were wrong, I sympathize with the difficult position you're in (I've spent about 20 years championing a hypothesis that almost everyone thinks is wrong). I also said that what matters in science isn't whether we make mistakes (we all do) but how we deal with them, and that I think you're handling the situation well.
I feel particularly bad about the 'not calm and confident' quote, because in fact your press conference was very well done. I meant this statement to only emphasize that women in science know that they're being judged harshly, but instead I came across as someone doing precisely that.
Sorry,
Rosie Redfield
p.s. to everyone else: I don't want this post to become a place to debate speaking styles so I'm going to close comments (or delete them if I can't figure out how to close them). My apologies to the four people who've already commented, as I'm about to delete your comments.
Dear Dr. Wolfe-Simon,
I'm emailing you to apologize for quotes from me that have just appeared on the ABC News website. I wasn't misquoted, but some of the things I said in a phone interview yesterday morning came across more harshly than I had intended.
I told the interviewer that, even though I think your conclusions were wrong, I sympathize with the difficult position you're in (I've spent about 20 years championing a hypothesis that almost everyone thinks is wrong). I also said that what matters in science isn't whether we make mistakes (we all do) but how we deal with them, and that I think you're handling the situation well.
I feel particularly bad about the 'not calm and confident' quote, because in fact your press conference was very well done. I meant this statement to only emphasize that women in science know that they're being judged harshly, but instead I came across as someone doing precisely that.
Sorry,
Rosie Redfield
p.s. to everyone else: I don't want this post to become a place to debate speaking styles so I'm going to close comments (or delete them if I can't figure out how to close them). My apologies to the four people who've already commented, as I'm about to delete your comments.
Back to blogging as usual
But I can't resist first posting this fabulous photo-mashup of a face-off between myself and Felisa Wolfe-Simon (from Gizmodo):
 Now, where was I? Last week's transformation experiment worked quite well. I didn't get nearly as many NovR and NalR transformants as I had expected, and some of the plates had contaminants, but I ended up with five independent pools containing between 5,000 and 50,000 independent clones each. So I diluted each pool to about 3 x 10^9 cells per ml, added glycerol, and froze four aliquots of each pool in nicely labelled tubes, all ready to FedEx to London (England, not Ontario) on Monday.
Now, where was I? Last week's transformation experiment worked quite well. I didn't get nearly as many NovR and NalR transformants as I had expected, and some of the plates had contaminants, but I ended up with five independent pools containing between 5,000 and 50,000 independent clones each. So I diluted each pool to about 3 x 10^9 cells per ml, added glycerol, and froze four aliquots of each pool in nicely labelled tubes, all ready to FedEx to London (England, not Ontario) on Monday.
Well, it's Thursday, and the cells are still in our freezer. Actually they're back in our freezer, because we shipped them out yesterday afternoon, thinking we had met all the requirements, and this morning FedEx brought them back. (But we still have to pay them for this on-shipment!)
The problem is that these are infectious bacteria, and the shipping agencies enforce very strict regulations about packaging and labelling. I think some central agency (government) must make the regulations, and packages must be prepared for shipping by a person who's taken a special training course in transport of hazardous goods. I probably should have realized this, but I didn't, and we had a really hard time finding the information we needed. (We haven't had to do this in the past - we've usually just sent people DNA rather than cells.)
Our Shipping and Receiving office originally told the RA to talk to Health and Safety, and Health and Safety didn't return the RA's call or her email. So on Tuesday I talked to FedEx, and then I called up Health and Safety and found out that (1) the shipper had to have taken a course; (2) it's not the kind of course you can just take in an hour online; (3) a technician in our building had taken the course and might help us. Then she told us that the manager of Shipping and Receiving had the training we needed, and sure enough he did. But it was too late to do it then.
He was busy Wednesday morning, but yesterday afternoon we took the frozen cells inside the special o-ring sealed plastic container inside the specially labelled cardboard box (we had received the container and box when someone else had shipped cells to us) inside the big styrofoam box. He got us the dry ice, checked all our paperwork and labels, signed the forms, and put his cell phone as the emergency 24-hr contact number. The RA had already set up the shipment online and printed out the waybill and commercial invoice (3 copies) and the Dangerous Shipment declaration (3 copies, each printed in colour on the lab next door's printer). She taped the styrofoam box shut and put in the special FedEx pickup place, and we both heaved sighs of relief.
But this morning the box came back. Once it had reached FedEx's central clearing house they'd gone over it with their 900-point checklist (I exaggerate only slightly) for hazardous-goods shipments, and it had failed. Not just one point - there were Xs in about ten of the boxes. We had forgotten to write the weight of the dry ice on the form. The shipping guy had forgot to sign one of the forms in one of the places, and to resign at somewhere a change had been made. Our styrofoam box needed to be inside a cardboard box, which must be labelled "OVERPACK". The Dangerous Shipment form must describe the contents with very precise wording. And it must not be completed by hand - of course we don't have a typewriter, and it's on a pdf form that can be typed into but not saved, so we'll ask the lab next door to let us borrow one of their computers as well as their colour printer - we can't just complete the form on one of our computers and send it to their printer because we're in different departments and thus our computers are on different networks.
Because the shipment will probably take longer than overnight to get to London, we didn't want to send it on a Thursday or Friday and risk having it sit around getting warm all weekend. So we're getting everything ready again to send it on Monday.

Well, it's Thursday, and the cells are still in our freezer. Actually they're back in our freezer, because we shipped them out yesterday afternoon, thinking we had met all the requirements, and this morning FedEx brought them back. (But we still have to pay them for this on-shipment!)
The problem is that these are infectious bacteria, and the shipping agencies enforce very strict regulations about packaging and labelling. I think some central agency (government) must make the regulations, and packages must be prepared for shipping by a person who's taken a special training course in transport of hazardous goods. I probably should have realized this, but I didn't, and we had a really hard time finding the information we needed. (We haven't had to do this in the past - we've usually just sent people DNA rather than cells.)
Our Shipping and Receiving office originally told the RA to talk to Health and Safety, and Health and Safety didn't return the RA's call or her email. So on Tuesday I talked to FedEx, and then I called up Health and Safety and found out that (1) the shipper had to have taken a course; (2) it's not the kind of course you can just take in an hour online; (3) a technician in our building had taken the course and might help us. Then she told us that the manager of Shipping and Receiving had the training we needed, and sure enough he did. But it was too late to do it then.
He was busy Wednesday morning, but yesterday afternoon we took the frozen cells inside the special o-ring sealed plastic container inside the specially labelled cardboard box (we had received the container and box when someone else had shipped cells to us) inside the big styrofoam box. He got us the dry ice, checked all our paperwork and labels, signed the forms, and put his cell phone as the emergency 24-hr contact number. The RA had already set up the shipment online and printed out the waybill and commercial invoice (3 copies) and the Dangerous Shipment declaration (3 copies, each printed in colour on the lab next door's printer). She taped the styrofoam box shut and put in the special FedEx pickup place, and we both heaved sighs of relief.
But this morning the box came back. Once it had reached FedEx's central clearing house they'd gone over it with their 900-point checklist (I exaggerate only slightly) for hazardous-goods shipments, and it had failed. Not just one point - there were Xs in about ten of the boxes. We had forgotten to write the weight of the dry ice on the form. The shipping guy had forgot to sign one of the forms in one of the places, and to resign at somewhere a change had been made. Our styrofoam box needed to be inside a cardboard box, which must be labelled "OVERPACK". The Dangerous Shipment form must describe the contents with very precise wording. And it must not be completed by hand - of course we don't have a typewriter, and it's on a pdf form that can be typed into but not saved, so we'll ask the lab next door to let us borrow one of their computers as well as their colour printer - we can't just complete the form on one of our computers and send it to their printer because we're in different departments and thus our computers are on different networks.
Because the shipment will probably take longer than overnight to get to London, we didn't want to send it on a Thursday or Friday and risk having it sit around getting warm all weekend. So we're getting everything ready again to send it on Monday.
Interested in doing research in my lab?
I'm taking advantage of this barrage of visitors to spread the word that my research group has openings for both a graduate student (M.Sc. or Ph.D.) and a postdoctoral fellow. We investigate the molecular biology and evolution of bacteria DNA uptake; you can read more about us at our home page, and find the details of our current research plans in our latest grant proposals.
My Letter to Science
(If you're looking for the long post I wrote on Saturday, the one that started the controversy about the Wolfe-Simon arsenic bacteria paper by describing all the problems I found, it's here.)
Below is the text of my formal Letter to Science about the Wolfe-Simon paper. Letter submissions are supposed to be limited to 300 words (this is a bit over at 371), so I'm only bringing up the issues concerning contamination. This is an improved version that incorporates many of the suggestions provided in the comments below. So some of the comments (in the first 10-15) won't make sense any more.
Because this paper has LOTS of other problems, it would be great if many other researchers could also submit Letters and Technical Comments (limit of 1000 words, peer-reviewed). Here's a link to the Instructions for Authors entry page. I won't mind if your Letter gets accepted and mine doesn't.
Here's mine:
Below is the text of my formal Letter to Science about the Wolfe-Simon paper. Letter submissions are supposed to be limited to 300 words (this is a bit over at 371), so I'm only bringing up the issues concerning contamination. This is an improved version that incorporates many of the suggestions provided in the comments below. So some of the comments (in the first 10-15) won't make sense any more.
Because this paper has LOTS of other problems, it would be great if many other researchers could also submit Letters and Technical Comments (limit of 1000 words, peer-reviewed). Here's a link to the Instructions for Authors entry page. I won't mind if your Letter gets accepted and mine doesn't.
Here's mine:
Wolfe-Simon et al. (1) meticulously eliminated contamination of the reagents and equipment used in their elemental analyses, but they made much less effort to eliminate contamination in their biological samples.
The reagents used for the culture media were not pure. The 3.1 µM PO4 contaminating the As+/P- medium provided enough P for all of the cell growth seen in this medium, using the authors’ estimate of 7.5x106 atoms of P per genome and the generous assumption that phosphate-starved cells use 90% of their P for molecules other than DNA (2). This calculation (not done by the authors) obviates their hypothesis that the cells could only grow by replacing P with As.
An independent contamination problem is the omission of standard DNA purification steps when testing for As in DNA (2). Contamination is typical in DNA/RNA pellets produced by ethanol precipitation of the aqueous phases from phenol:chloroform extractions. This is partly because this fraction contains most of the small molecules from the cytoplasm (contrary to the authors’ assertion), which are often less soluble in 70% ethanol than in water. Pellets are also typically contaminated with small amounts of the ethanol supernatant. Yet the usual step of washing the pellets was omitted, and the dried pellets were simply resuspended in water and loaded on an agarose gel.
Most surprisingly, the chromosomal DNA fractions (boxed in Fig. 2A) were not purified from the gel slices (a standard ten-minute procedure). Instead the authors simply dried the gel slices and assayed them. Not only does this bring in any contaminants present in the gel, but since each gel slice would have contained at least 1 mg of agarose (100 mg of 1% agarose gel), and each DNA band no more than 1 µg of DNA, at least 99.9% of the carbon in these samples would have come from the agarose, not the DNA. No correction can be made for the agarose-derived C because the actual amounts of DNA and agarose are not known. Omission of the gel-removal step for these critical samples is surprising because the authors did use it in preparing the rDNA fragments they sequenced for their phylogenetic analysis.
1. Wolfe-Simon F, Blum JS, Kulp TR, Gordon GW, Hoeft SE, Pett-Ridge J, Stolz JF, Webb SM, Weber PK, Davies PC, Anbar AD, & Oremland RS (2010). Science Express. PMID: 21127214
2. W. Makino, J. Cotner, R. Sterner, J. Elser, Funct Ecol 17,121 (2003).
3. J. Sambrook, D. W. Russell. Molecular Cloning, A Laboratory Manual. 3rd Ed. Cold Spring Harbor Press, New York 2001.
Subscribe to:
Posts (Atom)