














I repeated the fraction-competent assay on log-phase murE749 hypercompetent cells, as I said I should in the previous post. I did a very thorough and well-controlled experiment, but the results tell me that this isn't a very reliable measure of how much the cells in the culture differ in their competence.
I grew the cells at low density in rich medium for about 3.5 hours, so they would all be growing exponentially (in log phase). I added MAP7 DNA to the cells, let them grow for 15 minutes, and added DNase I to prevent continuing DNA uptake. I then let the cells continue growing for another 1.5 hours, to allow all the antibiotic resistance alleles to be fully expressed. Then I diluted the culture and spread the cells on agar plates containing different antibiotics, singly or in pairwise combinations.
MAP7 DNA contains point mutations causing resistance to 7 different antibiotics, but I only selected for 4 of them in this experiment: novobiocin (nov), kanamycin (kan), spectinomycin (spc) and nalidixic acid (nal). The nov and kan alleles are close together on the chromosome, so I didn't select for those two together, but I selected for nov+spc, noc+nal, nal+spc, kan+nal, and kan+spc. These combinations gave me 5 different measures of fraction competent.
The nal allele gave a low transformation frequency on its own (4.9x10^-4), and all 3 of the combinations that included nal gave low estimates of fraction competent: 0.06, 0.08 and 0.09. The other two combinations gave higher estimates: 0.28 and 0.58.
That's a ten-fold range of the estimates. Practically, the difference between 0.06 of the cells being competent and 0,58 being competent is enormous, but the fraction competent assays can't tell the difference.
A former student had proposed developing a fluorescent reporter-gene assay that would let us look at cells under the microscope and count the ones that had turned on their competence genes. I still think it would be a big pain, largely because the cells are so small, but maybe the recent improvements in reporter molecules and in microscopy now make this a good idea.
Field of Science
-
-
Change of address1 year ago in Variety of Life
-
Change of address1 year ago in Catalogue of Organisms
-
-
Earth Day: Pogo and our responsibility1 year ago in Doc Madhattan
inconclusive fraction-competent results
A few posts ago I described surprising results from an experiment measuring the fractions of the cells in different cultures that were competent. Here they are again:


Conditions were a bit different this time. First, the KW20 (wildtype) culture had not been induced to maximum competence this time - these cells were approaching stationary phase and are 50-100-times less comepetent. Second, this time I remembered to give all the cultures 60 minutes in rich medium before plating to allow expression fo the spectinomycin resistance.
Including expression time doesn't appear to have significantly changes the transformation frequencies for SpcR selected alone, but it substantially increased the double transformation frequencies (NovR SpcR), and this reduced the apparent fraction competent to below 1.0.
So this new experiment clarifies why I got the anomalously high FC in the previous experiment. Unfortunately it doesn't address the reason I wanted to measure FC in the hypercompetent mutants in the first place. The question arose from the analysis of the ∆HI0659 mutant's growth rates. In this experiment). I had found that cells carrying the HI0659 mutant grew normally.
I did this experiment because I wanted to find out whether unopposed expression of the HI0660 'toxin' harms cells (killing them or inhibiting their growth). In the ∆HI0659 cells the HI0660 'toxin' is induced but not opposed by the HI0659 'antitoxin' when competence genes are on. Because competence genes aren't normally on in growing cells anyway, I had tested growth in two hypercompetent mutant backgrounds, sxy-1 and murE749 - this was normal too.
The fraction competent experiments were intended to test whether most of the cells in the hypercompetent cultures has their competence genes on - if not then we might not see a dramatic growth difference even if the toxin does harm cells. But I foolishly did them with cells approaching stationary phase, when I should have done them with cells in exponential growth. I don't need to bother doing this for the sxy-1 mutant, since I already know that only a small fraction of its cells are competent then (I did this experiment years ago), but I should test it for murE749.

And here are the new results:

Including expression time doesn't appear to have significantly changes the transformation frequencies for SpcR selected alone, but it substantially increased the double transformation frequencies (NovR SpcR), and this reduced the apparent fraction competent to below 1.0.
So this new experiment clarifies why I got the anomalously high FC in the previous experiment. Unfortunately it doesn't address the reason I wanted to measure FC in the hypercompetent mutants in the first place. The question arose from the analysis of the ∆HI0659 mutant's growth rates. In this experiment). I had found that cells carrying the HI0659 mutant grew normally.
I did this experiment because I wanted to find out whether unopposed expression of the HI0660 'toxin' harms cells (killing them or inhibiting their growth). In the ∆HI0659 cells the HI0660 'toxin' is induced but not opposed by the HI0659 'antitoxin' when competence genes are on. Because competence genes aren't normally on in growing cells anyway, I had tested growth in two hypercompetent mutant backgrounds, sxy-1 and murE749 - this was normal too.
The fraction competent experiments were intended to test whether most of the cells in the hypercompetent cultures has their competence genes on - if not then we might not see a dramatic growth difference even if the toxin does harm cells. But I foolishly did them with cells approaching stationary phase, when I should have done them with cells in exponential growth. I don't need to bother doing this for the sxy-1 mutant, since I already know that only a small fraction of its cells are competent then (I did this experiment years ago), but I should test it for murE749.
hfq knockout results
I've now examined the effects of knocking out the small RNA-regulating protein Hfq under a wide range of conditions, testing our hypothesis that it regulates competence by helping unfold the translation-inhibiting stem of sxy mRNA.
In my previous experiment I found that the hfq knockout (∆hfq) causes a ten-fold decrease in transformation, both during growth in rich medium and after transfer to the starvation medium MIV. This time I also tested the mutation in combination with either of two hypercompetence-causing mutations (sxy-1 and murE749), and under culture conditions. The reasoning was that if ∆hfq's transformation defect is due to a defect in sxy translation, it should be reduced or eliminated by the sxy-1 mutation, which we know destabilizes the RNA stem. Seeing a similar effect of the murE749 mutation might suggest that this mutation also acts by destabilizing an RNA pairing structure, perhaps the same sxy mRNA stem.
Here are the results.

Starting from the bottom up: In the competence-inducing medium MIV we see the same ~10-fold defect in the wildtype background but no defect in the sxy-1 or murE749 backgrounds. This supports the above hypothesis and suggests that the murE749 mutation also acts by disrupting RNA pairing.
We think that transfer to MIV medium causes two events that together cause expression of the competence genes: (i) cAMP levels go up, and (ii) the mRNA stem no longer blocks translation of sxy mRNA into Sxy protein. Simply adding cAMP to log-phase cells induces only a low level of competence, since the mRNA stem continues to block its translation. This predicts that adding cAMP to hfq mutants will give 10-fold lower competence, but instead we see that competence is nearly normal in the wildtype background and fully normal in the sxy-1 or murE749 backgrounds. This suggests that ∆hfq's competence defect is not duo to a defect in destabilizing the sxy mRNA stem, but instead to an effect on intracellular cAMP levels.
In late-log cells (in rich medium) we think that the low-level competence normally observed is due to a spontaneous increase in cAMP levels, not to destabilization of the sxy mRNA stem. But the experiment saw a larger-than-expected defect in the wildtype background, a ~10-fold defect in the sxy-1 background, and no defect in the murE749 background. I don't know what to make of this.
The final condition was 'overnight cultures' - cultures that grew to maximum density and remained at 37°C on the roller wheel until morning. The hfq+ and ∆hfq cultures in the wildtype background gave no transformants at all, but both hypercompetent backgrounds showed much stronger competence defects than under other conditions (>100-fold). However this could be an artefact of the cessation of growth on expression of the novobiocin resistance allele.
Overall, what should we conclude? I find the cAMP results to be the most compelling; they strongly suggest that our hypothesis is wrong; Hfq does not contribute to the translatability of sxy mRNA.
In my previous experiment I found that the hfq knockout (∆hfq) causes a ten-fold decrease in transformation, both during growth in rich medium and after transfer to the starvation medium MIV. This time I also tested the mutation in combination with either of two hypercompetence-causing mutations (sxy-1 and murE749), and under culture conditions. The reasoning was that if ∆hfq's transformation defect is due to a defect in sxy translation, it should be reduced or eliminated by the sxy-1 mutation, which we know destabilizes the RNA stem. Seeing a similar effect of the murE749 mutation might suggest that this mutation also acts by destabilizing an RNA pairing structure, perhaps the same sxy mRNA stem.
Here are the results.

We think that transfer to MIV medium causes two events that together cause expression of the competence genes: (i) cAMP levels go up, and (ii) the mRNA stem no longer blocks translation of sxy mRNA into Sxy protein. Simply adding cAMP to log-phase cells induces only a low level of competence, since the mRNA stem continues to block its translation. This predicts that adding cAMP to hfq mutants will give 10-fold lower competence, but instead we see that competence is nearly normal in the wildtype background and fully normal in the sxy-1 or murE749 backgrounds. This suggests that ∆hfq's competence defect is not duo to a defect in destabilizing the sxy mRNA stem, but instead to an effect on intracellular cAMP levels.
In late-log cells (in rich medium) we think that the low-level competence normally observed is due to a spontaneous increase in cAMP levels, not to destabilization of the sxy mRNA stem. But the experiment saw a larger-than-expected defect in the wildtype background, a ~10-fold defect in the sxy-1 background, and no defect in the murE749 background. I don't know what to make of this.
The final condition was 'overnight cultures' - cultures that grew to maximum density and remained at 37°C on the roller wheel until morning. The hfq+ and ∆hfq cultures in the wildtype background gave no transformants at all, but both hypercompetent backgrounds showed much stronger competence defects than under other conditions (>100-fold). However this could be an artefact of the cessation of growth on expression of the novobiocin resistance allele.
Overall, what should we conclude? I find the cAMP results to be the most compelling; they strongly suggest that our hypothesis is wrong; Hfq does not contribute to the translatability of sxy mRNA.
Choosing a journal for your manuscript
Listening to Bruce Dancik's #CSPPubTour12 talk yesterday about choosing a journal and submitting your manuscript got me thinking about issues he didn't emphasize. I started with a few, but my list keeps getting longer and longer:
Access to your article: Does the journal provide immediate open access for all its papers? Is this an option, for an extra charge? Open
access after 6 months or a year? Subscription
only? If subscription access, how widely
is it subscribed to?
Cost of publishing: Are there page charges? Optional or required publication charges for open access? Charges for colour figures (only an issue for print journals)?
Limits on article length: No limit? Very
tight? Charges for extra pages?
Likelihood of acceptance: Do your subject, approach and results fit the mandate of the
journal (is yours the kind of manuscript they’re looking for)?
Prestige for your CV: How good is the journal's reputation? How high is its impact factor?
Prestige for journalists: Are papers from this journal often reported in the mainstream media?
Readership:
Does the journal cover a specialized topic or a broad area of science? Which kind of audience are you writing for?
Ease of finding for readers: Is the journal indexed by everything? How well can you use keywords in the
title and abstract to bring in readers from Google Scholar and other search
engines?
Cost of publishing: Are there page charges? Optional or required publication charges for open access? Charges for colour figures (only an issue for print journals)?
Online supplementary materials: Does the journal host these? What are the limitations?
Copyright and licensing: Must you sign away your rights? Can others reuse your material (e.g. use your figures in teaching)?
Turnaround time:
Rapid pre-screening? Total
time from submission to publication?
Online early access?
Is this an ethical publisher? Elsevier? Other
for-profit? Society journal? Predatory publisher (see Beall's list)?
Type of publication:
Online-only? Print edition
only? Both?
Tiresomeness of Instructions to Authors: Will getting your figures into the obscure required format force you to spend $500 on the full version of Photoshop?
Might the journal highlight your paper? Does it include a News and Views or other section that highlights some papers in each issue?
Relative activities of comM from strains Rd and NP
A month or so ago I described four experiments I wanted to do. I've now done the last of them, testing whether the comM gene of strain NP could be partly responsible for that strain's 100-fold lower transformability.
...and the answer appears to be... NO (with one qualification).
The experiment was to compare the abilities of plasmid-borne Rd and NP comM genes to restore full competence to Rd cells whose chromosomal comM gene had been deleted. The RA made all the strains for me - all I had to do was measure their transformation frequencies after fully inducing competence by transfer to starvation medium. The first four bars in the graph below show the results:

Both comM alleles restore normal competence to the Rd knockout when cloned in the forward orientation but not when cloned in the reverse orientation. I think each insert has its own CRP-S promoter, but the plasmid also carries the E. coli lacZ promoter which, I suspect, interferes with expression of inserts whose promoters face in the reverse orientation.
The motivation behind this experiment was a recombinant strain identified by an undergraduate who was working with the postdoc. This strain transforms only 10% as well as Rd; it has been sequenced and we know it contains a single 40 kb segment of NP sequence. The only known competence gene in this 40 kb segment is comM, so they hypothesized that a partially defective NP comM might be responsible for the recombinant's 10-fold reduced competence and partly responsible for NP's 100-fold lower competence. This new result suggests that this hypothesis is wrong.
The qualification is that strong expression from a plasmid could mask lower expression or catalytic activity of NP's comM gene. The gold standard experiment would be to replace the Rd chromosomal allele with the NP version and vice versa. For various technical reasons we haven't been able to do this yet.
...and the answer appears to be... NO (with one qualification).
The experiment was to compare the abilities of plasmid-borne Rd and NP comM genes to restore full competence to Rd cells whose chromosomal comM gene had been deleted. The RA made all the strains for me - all I had to do was measure their transformation frequencies after fully inducing competence by transfer to starvation medium. The first four bars in the graph below show the results:

The motivation behind this experiment was a recombinant strain identified by an undergraduate who was working with the postdoc. This strain transforms only 10% as well as Rd; it has been sequenced and we know it contains a single 40 kb segment of NP sequence. The only known competence gene in this 40 kb segment is comM, so they hypothesized that a partially defective NP comM might be responsible for the recombinant's 10-fold reduced competence and partly responsible for NP's 100-fold lower competence. This new result suggests that this hypothesis is wrong.
The qualification is that strong expression from a plasmid could mask lower expression or catalytic activity of NP's comM gene. The gold standard experiment would be to replace the Rd chromosomal allele with the NP version and vice versa. For various technical reasons we haven't been able to do this yet.
A rotation student replaced the recombinants NP comM allele with the knocked-out Rd version - this reduced its transformation frequency by another 10-fold, about the same as the TF of a simple RD ∆comM strain.
I had tried to transform a ∆comM strain of NP with the Rd comM allele, but the ∆comM mutant the RA made was, unexpectedly, completely non-transformable. She has now given me another NP ∆comM isolate to test. That's the last column in the chart; the red star indicates no transformant colonies at all, so this isolate too is completely non-transformable. This result is consistent with the similarity of the Rd and NP comM plasmid results; they both suggest that the NP comM gene is fully functional, and that some other difference(s) must be responsible for its lower transformability.
So why does the recombinant have lower transformability? Before designing any more experiments I need to be better able to think about the relative chromosomal locations of the various selectable markers and competence genes we're interested in. To this end I'm having our work-study student take a break from glassware-washing and media preparation to do what she calls an 'arts and crafts' project - making us a poster showing the locations of all these genes drawn on a circular chromosome. I just need to give her a list of the genes/ locations to include on her poster.
He takes after me!
Here's a photo of my nephew's science project. He represented the features of the eukaryote cell using cake and candy. The nucleus is a peanut-butter cup, with a jaw-breaker nucleolus. He got a mark of 100%.


A new twist on the fraction-competent problem
On Sunday I attempted to measure what fraction of the cells in some cultures were competent (able to take up DNA fragments and recombine them into their chromosome). I've previously written about this kind of analysis here and here.
Usually when I do this kind of experiment I find that only some cells were competent (anywhere from about 10% to about 50%). But this time I got a very different result, and I don't know why.
I was testing three strains - wildtype cells (strain KW20) and two hypercompetent mutant derivatives, carrying the sxy1 and murE749 mutations. Both KW20 and sxy1 have been tested several times before; mutrE749 hasn't.
The procedure is simple. Transform cells with MAP7 chromosomal DNA and select for two antibiotic resistance mutations located far apart on the chromosome (so they won't ever be carried on the same DNA fragment). Select for each resistance separately and for the two together (double-transformants). Count the resulting colonies and calculate the transformation frequency for each resistance separately and for the double-transformants.
Calculate the fraction competent as the product of the two single-mutation transformation frequencies divided by the double-transformant frequency and by a fudge factor somewhere between 2 and 4.
The fudge factor incorporates two sub-factors accounting for different effects. The first effect is the chance that a single cell took up and recombined two DNA fragments, each containing one of the mutations, but that the incoming DNAs recombined with different strands so that, when the cell divided, one daughter cell got one mutation and the other got the other. This factor is complicated by the unknown effects of mismatch repair, but on average 2 is the appropriate value. The second effect is whether the cells did any cell divisions before they were placed on the agar to grow into colonies. If they did, then cells transformed by a single mutation will have given rise to one resistant colony and one sensitive colony. This factor should have a value of 2 if all the cells had time to divide before plating (if, for example, they needed time to express the antibiotic resistance), by less than 2 if only some did, and by 1 if the cells were plated immediately after transformation. In my experiment I didn't allow any expression time* so this second factor should be closer to 1 than 2. For simplicity I'll use a complete fudge factor of 3.
Here are the calculations:
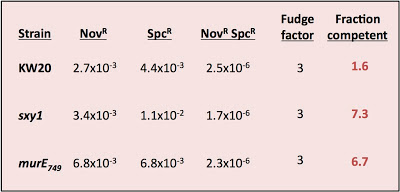
What's wrong with these nice numbers? The fraction competent should ALWAYS be less than 1; that's why it's called a fraction!
Might my assumptions be invalid? This analysis requires that cells be able to take up more than one fragment of DNA, and that taking up one fragment does not affect the probability of taking up another fragment. Because previous experiments have always given values less than 1, we've been assuming that this requirement is met. But this new result only makes sense if many of the cells could only take up one fragment of DNA.
Might my DNA or plates be faulty? The problem isn't due to using some new DNA prep with different properties; I used a fresh tube of the same MAP7 DNA stock we've been using for years. Could it be because I forgot to give the cells some expression time after DNA uptake? Might the lack of expression time reduced the numbers of double transformants (SpcR NovR) much more than it reduced the numbers of single SpcR transformants? But lack of expression time doesn't appear to have been a problem, since I got at least as many SpcR transformants as NovR transformants. The DNase I stock could be a problem. The transformation reactions are stopped after 15 minutes by adding DNase I to degrade the remaining DNA - a few days ago I tested the DNase I stock, by adding it to the DNA 5 minutes before I added the cells, and found that it wasn't very effective - the residual transformation frequency was still quite high. This means that some cells might have taken up DNA while they were on the plate, but since this would only exacerbate the expression-time problem it shouldn't have been a big factor.
I only did this experiment because it would help us interpret the lack of effect of the HI0659 knockout on growth rates (in the experiment I described yesterday). If most cells weren't competent even in the hypercompetence mutants, then not seeing a growth defect in the culture doesn't meen that the competent cells didn't experience a growth defect. But this weird result means I need to do more experiments to figure out the reason for the discrepancy with previous results.
Usually when I do this kind of experiment I find that only some cells were competent (anywhere from about 10% to about 50%). But this time I got a very different result, and I don't know why.
I was testing three strains - wildtype cells (strain KW20) and two hypercompetent mutant derivatives, carrying the sxy1 and murE749 mutations. Both KW20 and sxy1 have been tested several times before; mutrE749 hasn't.
The procedure is simple. Transform cells with MAP7 chromosomal DNA and select for two antibiotic resistance mutations located far apart on the chromosome (so they won't ever be carried on the same DNA fragment). Select for each resistance separately and for the two together (double-transformants). Count the resulting colonies and calculate the transformation frequency for each resistance separately and for the double-transformants.
Calculate the fraction competent as the product of the two single-mutation transformation frequencies divided by the double-transformant frequency and by a fudge factor somewhere between 2 and 4.
The fudge factor incorporates two sub-factors accounting for different effects. The first effect is the chance that a single cell took up and recombined two DNA fragments, each containing one of the mutations, but that the incoming DNAs recombined with different strands so that, when the cell divided, one daughter cell got one mutation and the other got the other. This factor is complicated by the unknown effects of mismatch repair, but on average 2 is the appropriate value. The second effect is whether the cells did any cell divisions before they were placed on the agar to grow into colonies. If they did, then cells transformed by a single mutation will have given rise to one resistant colony and one sensitive colony. This factor should have a value of 2 if all the cells had time to divide before plating (if, for example, they needed time to express the antibiotic resistance), by less than 2 if only some did, and by 1 if the cells were plated immediately after transformation. In my experiment I didn't allow any expression time* so this second factor should be closer to 1 than 2. For simplicity I'll use a complete fudge factor of 3.
* In retrospect I should have allowed expression time, because one of the markers I was selecting for is spectinomycin resistance, which we think of as needing an hour's expression time. But I forgot and, rather surprisingly, still got tons of transformants.
Here are the calculations:

Might my assumptions be invalid? This analysis requires that cells be able to take up more than one fragment of DNA, and that taking up one fragment does not affect the probability of taking up another fragment. Because previous experiments have always given values less than 1, we've been assuming that this requirement is met. But this new result only makes sense if many of the cells could only take up one fragment of DNA.
Might my DNA or plates be faulty? The problem isn't due to using some new DNA prep with different properties; I used a fresh tube of the same MAP7 DNA stock we've been using for years. Could it be because I forgot to give the cells some expression time after DNA uptake? Might the lack of expression time reduced the numbers of double transformants (SpcR NovR) much more than it reduced the numbers of single SpcR transformants? But lack of expression time doesn't appear to have been a problem, since I got at least as many SpcR transformants as NovR transformants. The DNase I stock could be a problem. The transformation reactions are stopped after 15 minutes by adding DNase I to degrade the remaining DNA - a few days ago I tested the DNase I stock, by adding it to the DNA 5 minutes before I added the cells, and found that it wasn't very effective - the residual transformation frequency was still quite high. This means that some cells might have taken up DNA while they were on the plate, but since this would only exacerbate the expression-time problem it shouldn't have been a big factor.
I only did this experiment because it would help us interpret the lack of effect of the HI0659 knockout on growth rates (in the experiment I described yesterday). If most cells weren't competent even in the hypercompetence mutants, then not seeing a growth defect in the culture doesn't meen that the competent cells didn't experience a growth defect. But this weird result means I need to do more experiments to figure out the reason for the discrepancy with previous results.
the HI0660 'toxin' doesn't affect cell growth or survival
My last experiment showed that HI0660 encodes a 'toxin' of some sort, which prevents transformation when induced in the absence of the antitoxin encoded by HI0659. So yesterday I did detailed growth curves for cultures carrying the HI0659 knockout and either of our hypercompetence mutations.
The logic is that neither HI0660 or HI0659 will be expressed during growth in wildtype cultures, because the competence genes are not induced then. They're weakly induced at the end of growth, but strongly induced only when cells are starved or in the presence of the hypercompetence mutations. This means that, if HI0660 does kill cells or prevent growth, this effect will best be seen in the presence of the hypercompetence mutations.
The results show no evidence of significant growth or survival differences due to unopposed expression of HI0660. Each line in the two graphs below shows the mean for 7 replicate wells of the same inoculum. The upper graph used inocula that were of single small colonies diluted into 10 ml medium. The lower graph used inocula that were 1/1000 dilutions of overnight cultures into medium.
This is an interesting result, because it suggests that HI0660 acts directly on DNA uptake, not by killing cells or interfering with their growth. Because homologs of HI0660 are known to act by inactivating specific mRNAs, we may have to use RNA-seq to identify its mode of action.


The logic is that neither HI0660 or HI0659 will be expressed during growth in wildtype cultures, because the competence genes are not induced then. They're weakly induced at the end of growth, but strongly induced only when cells are starved or in the presence of the hypercompetence mutations. This means that, if HI0660 does kill cells or prevent growth, this effect will best be seen in the presence of the hypercompetence mutations.
The results show no evidence of significant growth or survival differences due to unopposed expression of HI0660. Each line in the two graphs below shows the mean for 7 replicate wells of the same inoculum. The upper graph used inocula that were of single small colonies diluted into 10 ml medium. The lower graph used inocula that were 1/1000 dilutions of overnight cultures into medium.
This is an interesting result, because it suggests that HI0660 acts directly on DNA uptake, not by killing cells or interfering with their growth. Because homologs of HI0660 are known to act by inactivating specific mRNAs, we may have to use RNA-seq to identify its mode of action.


Woo-hoo!! A hypothesis proved correct!
Last spring I came up with a far-fetched hypothesis to explain the phenotypes of two of our competence-gene knockouts,HI0569 (competence eliminated) and HI0660 (normal competence). I proposed that HI0660n encoded a 'toxin' that prevents competence or kills cells expressing it, and that HI0659 encodes an antitoxin that protects cells from the actions of HI0660. You can read all about it here.
Yesterday I finally was able to do the critical experiment, testing the competence phenotype of cells with both genes knocked out. It's normal!
And here's the data:


Yesterday I finally was able to do the critical experiment, testing the competence phenotype of cells with both genes knocked out. It's normal!
And here's the data:

The asterisk on the HI0659 column indicates that this is a 'less than' data point, since there were no transformant colonies on any of the plates.
This result confirms that HI0660 does something that COMPLETELY prevents transformation, and that HI0659's job is to prevent HI0660 from doing whatever it does.

hfq results
Yes indeed, hfq is needed for full competence development. The mutant grows as well as its wild-type parent (top graph) but develops about ten-fold lower competence (lower graph).

Next step: Make DNA from the mutant and use it to transform the hfq knockout into the hypercompetent mutants. Then I'll test the effect of the hfq knockout on their competence. If Hfq avcts directly on the sxy mRNA stem that regulates translation, I expect the mutants to be unaffected by loss of Hfq because their translation is not limited by the stem.
As a check that Hfq's effects aren't due to indirect effects on cAMP levels or CRP activity, I'll also test the effect of the knockout in wildtype and hypercompetent cells with added cAMP.
What about effects on the murE hypercompetence mutants? I'll test that, but I'm not sure how to interpret different results...
I'm finally testing a hfq mutant
The RA's gone off on a short but well-earned trip to the sun, and before she left she made the H. influenzae hfq knockout I've been waiting for. So yesterday, after clicking 'Submit' on our NSERC grant proposal, I did competence time courses of wildtype cells and the mutant, hoping to see a difference in transformation frequencies (preferably a decrease).
You can read all the background in this post from last March. And here's a hexameric structure for Hfq (from Wikipedia):

The colonies are still too tiny to count, but it looks like the transformation frequency is down about 10-fold in the mutant!
Next step, make some DNA from the mutant so I can introduce the mutation into my hypercompetent mutant backgrounds.
You can read all the background in this post from last March. And here's a hexameric structure for Hfq (from Wikipedia):
The colonies are still too tiny to count, but it looks like the transformation frequency is down about 10-fold in the mutant!
Next step, make some DNA from the mutant so I can introduce the mutation into my hypercompetent mutant backgrounds.
Of HQPs and KM plans
I don't know why this latest round of grant proposal writing sapped my drive to write blog posts. But the last of the three proposals (to NSERC, Canada's equivalent of NSF) is pretty much done, and I'm turning my mind to doing some experiments.

But first, some NSERC acronyms:
NSERC: (I have to check every time...) Canada's Natural Sciences and Engineering Research Council.
HQP: NSERC is obsessed with training of 'highly qualified personnel'. Fully one third of the proposal's score is determined by our past production of HQP and our plans for producing HQPs from whatever trainees our successful grant might support. Both have to be meticulously documented, on penalto of having your proposal dumped in the recycling bin.
KM: I had to Google this one. The 'NSERC Tips and Tricks' document kindly provided by our research services people recommends that our research plan includes comments on open access (for sure!), and follows this with:

But first, some NSERC acronyms:
NSERC: (I have to check every time...) Canada's Natural Sciences and Engineering Research Council.
HQP: NSERC is obsessed with training of 'highly qualified personnel'. Fully one third of the proposal's score is determined by our past production of HQP and our plans for producing HQPs from whatever trainees our successful grant might support. Both have to be meticulously documented, on penalto of having your proposal dumped in the recycling bin.
KM: I had to Google this one. The 'NSERC Tips and Tricks' document kindly provided by our research services people recommends that our research plan includes comments on open access (for sure!), and follows this with:
If results are appropriate for open dissemination, what is your KM plan? If results are not appropriate, mention why.Google says that KM is knowledge mobilization (this cracked the post-doc up). So now my HQP training plan says that "Everyone is encouraged to keep an open research blog, providing both writing experience and knowledge mobilization."
Erudite Journals: Yet another predatory journal strategy
I just got an email invitation to publish in the Erudite Journal of Microbiology and Biodiversity. This is a group; they list ten other Erudite journals.
 Their publication fee is only $300, but they offer a special Language-Editing service for authors. For $150, they will edit your manuscript's writing (though not the equations), independent of its length, with a turnaround time of one week. After the editing is done you can decide whether to proceed with the submission.
Their publication fee is only $300, but they offer a special Language-Editing service for authors. For $150, they will edit your manuscript's writing (though not the equations), independent of its length, with a turnaround time of one week. After the editing is done you can decide whether to proceed with the submission.
At first I wondered whether I might be unfair in treating Erudite Journals as predatory. Maybe the publishers genuinely want to help researchers from non-English speaking countries publish their work. However, the Erudite Journal of Microbiology and Biodiversity has no Editor, and its archive has no papers. Neither does the Erudite Journal of Biotechnology, though it does claim that a first issue is coming soon (hopefully not before they find an Editor). Neither does the Erudite Journal of Law and Jurisprudence, or most of their other journals. The Erudite Journal of Business Administration does have an Editor, as do a couple of others, but the Erudite Journal of Social Science Research appears to be edited by a page full of advertisements.
Note added Sept. 24, 2013: I got another email, "Introducing Erudite Journals". Still no papers in the Erudite Journal of Microbiology and Biodiversity or the Erudite Journal of Law and Jurisprudence, but there's one in the Erudite Journal of Biotechnology! I don't see how they can possibly be making enough money to even pay for their website and spam.

At first I wondered whether I might be unfair in treating Erudite Journals as predatory. Maybe the publishers genuinely want to help researchers from non-English speaking countries publish their work. However, the Erudite Journal of Microbiology and Biodiversity has no Editor, and its archive has no papers. Neither does the Erudite Journal of Biotechnology, though it does claim that a first issue is coming soon (hopefully not before they find an Editor). Neither does the Erudite Journal of Law and Jurisprudence, or most of their other journals. The Erudite Journal of Business Administration does have an Editor, as do a couple of others, but the Erudite Journal of Social Science Research appears to be edited by a page full of advertisements.
Note added Sept. 24, 2013: I got another email, "Introducing Erudite Journals". Still no papers in the Erudite Journal of Microbiology and Biodiversity or the Erudite Journal of Law and Jurisprudence, but there's one in the Erudite Journal of Biotechnology! I don't see how they can possibly be making enough money to even pay for their website and spam.
I've already done Expt. C once; need to repeat
In my last post I described four experiments/series of experiments I wanted to do. One series (= Expt. C) I'd blogged about back in June; at that time I wrote that I was all set to do the experiments (strains frozen in log phase and ready to go). I hadn't blogged about any results, so I jumped to the conclusion that I hadn't yet done the experiments, but yesterday I discovered I had done them, with the results all neatly analyzed and graphed in my notebook.
Here are the results. The top graph shows the culture densities and the bottom graph shows the transformation frequencies. The purpose of the experiments was to find out if any of the cell-wall-peptide recycling mutations the 4 different colours of bars, plus the turquoise wildtype control) alter transformation frequency under conditions that do or don't induce competence. I had to control for culture density because this has a strong influence on competence induction.


Here are the results. The top graph shows the culture densities and the bottom graph shows the transformation frequencies. The purpose of the experiments was to find out if any of the cell-wall-peptide recycling mutations the 4 different colours of bars, plus the turquoise wildtype control) alter transformation frequency under conditions that do or don't induce competence. I had to control for culture density because this has a strong influence on competence induction.


The first two sets of bars are from the first experiment, testing cells in log phase (not inducing, left set of bars) or in log phase plus cyclic AMP (partially inducing, second set of bars). There's very little difference in culture density or transformation frequency between any of the mutants and the wildtype control. But the values for the log phase transformation frequencies (bottom graph, left set of bars) is not very reliable because the total numbers of antibiotic resistant colonies I counted were 1, 5, 1, 1, 4. To get better numbers I need to repeat this experiment using cells at a slightly higher density (OD = 0.2 rather than 0.1 (still log phase) and plating larger volumes of cells (using more plates).
I did the second experiment twice because the culture densities were too low the first time. There appear to be some (small) consistent differences in transformation frequency between the different mutants. But some of the numbers are still unreliable (too low, or too many colonies to count), so I need to do this experiment again too.
The problem with the low cell densities was partly because my plan of starting each experiment with cells that were already in log phase (grown and frozen in a previous experiment) didn't work very well. Apparently cells in log phase don't survive being frozen and thawed very well. I started each culture by thawing cells, diluting them into fresh medium, and checking the OD (measure of culture turbidity), but when I checked the OD an hour later it usually had gone down instead of increasing 3-4 fold. Apparently most of the cells contributing to the initial OD were not viable. So then I'd have to allow the culture an extra 2 or more hours for the surviving cells to reach the desired density, or close enough that I thought I could use them. And because these cells are the survivors of whatever trauma the freezing and thawing may have induced, they might not yet have settled down into a reproducible physiological state, which might have influenced their levels of competence.
So this time (first experiment repeat tomorrow, second one repeat on Tuesday) I'm going to allow lots of time for the cells to recover from freezing and get comfortably into log phase before I transform them.
Oops, RRResearch has become RRProposals andRRPublishing
It's been four months since I posted about an experiment! (I discovered this because I looked back through old posts to see what experiment I should start with.) I had one experiment (A below) underway, one (B below) planned (and blogged about ) but not actually started, and one (C below) that was waiting for the RA to make a mutant.
After consulting with the RA, here are some plans:
Experiment A: The goal is to make a H. influenzae mutant strain that has two genes knocked (HI0659 and HI0660). On its own, a HI0659 knockout eliminates competence; a HI0660 knockout has no effect. Because both genes have homologs in toxin-antitoxin systems, I hypothesize that HI0659's job is to prevent HI0660 from doing something toxic when it is induced in competent cells. This predicts that a double knockout will have normal competence.
I had been going to make the double mutant myself, while the RA was on leave, but now she's back she's got this underway. Next week she is going to create the double-mutant plasmid in E. coli (she has everything ready except the electro-competent cells) I'll then transform this mutant segment into H. influenzae and test competence, with both single mutants and wildtype cells as controls.
Experiment B: I want to carefully recheck the competence phenotypes of all our hypercompetent murE mutants, under several conditions. This is basically a long series of competence assays; I just need to streak out the various strains and get to work.
Experiment C: The secondary structure of the sxy gene's mRNA regulates its expression, and the Hfq protein contributes to gene regulation by helping small regulatory RNAs (sRNAs) find and bind to their target mRNAs. So we're going to make a H. influenzae hfq knockout and test its effect on competence. The RA is making the knockout - it's at the same stage as the double-mutant knockout described above. Once she's made the knockout in E. coli (next week), I'll transform it into H. influenzae and test competence.
Experiment D: I don't think I've ever done a blog post about this - it arises out of the post-doc's experiments transforming fragments of the clinical strain 86-028NP into the lab strain Rd and sequencing the recombinants. (He doesn't seem to have posted about it either.) 86-028NP transforms about 100-fold less well than Rd. None of the recombinants acquired the full transformation defect with their segments of 86-028NP DNA, but one of them transforms about tenfold worse than Rd.
The only known Rd competence gene acquired by this recombinant is comM, and the post-doc has hypothesized that its lower competence is due to replacement of the Rd comM allele with its 86-028NP homolog. ComM increases transformation frequencies by protecting incoming DNA strands from degradation in the cytoplasm. Knockouts have normal DNA uptake but about 50-fold lower transformation frequencies. (In other species the transformation defect is more severe.) Under the post-doc's hypothesis, the 86-028NP allele would be less active than its Rd homolog.

One way to test this is to restore the Rd comM allele to this transformant (replacing the one from 86-028NP but not the other 86-028NP sequences) - if differences in comM are responsible for the differences in transformation, this should increase transformation frequencies back to the Rd level. The RA has tried to do this but the construction didn't work - I may try it again. As an alternative she's put plasmids carrying the Rd or 86-28NP alleles into the recombinant strain, and I'm going to test these strains for differences in transformation frequency.
After consulting with the RA, here are some plans:
Experiment A: The goal is to make a H. influenzae mutant strain that has two genes knocked (HI0659 and HI0660). On its own, a HI0659 knockout eliminates competence; a HI0660 knockout has no effect. Because both genes have homologs in toxin-antitoxin systems, I hypothesize that HI0659's job is to prevent HI0660 from doing something toxic when it is induced in competent cells. This predicts that a double knockout will have normal competence.
I had been going to make the double mutant myself, while the RA was on leave, but now she's back she's got this underway. Next week she is going to create the double-mutant plasmid in E. coli (she has everything ready except the electro-competent cells) I'll then transform this mutant segment into H. influenzae and test competence, with both single mutants and wildtype cells as controls.
Experiment B: I want to carefully recheck the competence phenotypes of all our hypercompetent murE mutants, under several conditions. This is basically a long series of competence assays; I just need to streak out the various strains and get to work.
Experiment C: The secondary structure of the sxy gene's mRNA regulates its expression, and the Hfq protein contributes to gene regulation by helping small regulatory RNAs (sRNAs) find and bind to their target mRNAs. So we're going to make a H. influenzae hfq knockout and test its effect on competence. The RA is making the knockout - it's at the same stage as the double-mutant knockout described above. Once she's made the knockout in E. coli (next week), I'll transform it into H. influenzae and test competence.
Experiment D: I don't think I've ever done a blog post about this - it arises out of the post-doc's experiments transforming fragments of the clinical strain 86-028NP into the lab strain Rd and sequencing the recombinants. (He doesn't seem to have posted about it either.) 86-028NP transforms about 100-fold less well than Rd. None of the recombinants acquired the full transformation defect with their segments of 86-028NP DNA, but one of them transforms about tenfold worse than Rd.
The only known Rd competence gene acquired by this recombinant is comM, and the post-doc has hypothesized that its lower competence is due to replacement of the Rd comM allele with its 86-028NP homolog. ComM increases transformation frequencies by protecting incoming DNA strands from degradation in the cytoplasm. Knockouts have normal DNA uptake but about 50-fold lower transformation frequencies. (In other species the transformation defect is more severe.) Under the post-doc's hypothesis, the 86-028NP allele would be less active than its Rd homolog.

One way to test this is to restore the Rd comM allele to this transformant (replacing the one from 86-028NP but not the other 86-028NP sequences) - if differences in comM are responsible for the differences in transformation, this should increase transformation frequencies back to the Rd level. The RA has tried to do this but the construction didn't work - I may try it again. As an alternative she's put plasmids carrying the Rd or 86-28NP alleles into the recombinant strain, and I'm going to test these strains for differences in transformation frequency.
OK, about that Tawfik paper on arsenate resistance
Nice work. But I think it should be of interest only to people who care about the biochemistry of arsenic resistance, a set that doesn't include me.
GFAJ-s level of arsenate resistance is not all that exceptional. What was important about the Wolfe-Simon et al. paper was the claim that GFAJ-1 actively incorporates arsenic in place of phosphorus. We already know that this claim was an error (the sum of many errors) by the authors.
I'm returning my attention to the much more interesting question of whether bacteria have any functional parallel to the meiotic sex of eukaryotes.
GFAJ-s level of arsenate resistance is not all that exceptional. What was important about the Wolfe-Simon et al. paper was the claim that GFAJ-1 actively incorporates arsenic in place of phosphorus. We already know that this claim was an error (the sum of many errors) by the authors.
I'm returning my attention to the much more interesting question of whether bacteria have any functional parallel to the meiotic sex of eukaryotes.
Sunshine and benchwork = bliss?
I'm back... (I don't know why I haven't been posting while I've been grant-writing.)
Anyway, to start off easy, here's today's weather forecast:
 Nothing but sunshine for the next week. (Normally most days in October are rainy.) And nothing but sunshine for the past 2 months and more! Since July 24 Vancouver has had a total of only 8 mm of rain (normal is more than 100 mm). Anywhere else people would be worrying about the drought, and about climate change, but here we're just glorying in all the sunshine.
Nothing but sunshine for the next week. (Normally most days in October are rainy.) And nothing but sunshine for the past 2 months and more! Since July 24 Vancouver has had a total of only 8 mm of rain (normal is more than 100 mm). Anywhere else people would be worrying about the drought, and about climate change, but here we're just glorying in all the sunshine.
Two grant proposals have been submitted. You can get the CIHR one on the 'What we're planning' page of our website (link in the left sidebar); the CFC one will appear there soon. The first is a big one to CIHR (Canadian Institutes for Health Research, Canada's NIH), proposing to develop the information base and algorithm needed to predict transformational recombination in the respiratory tract. The second is a smaller one to Cystic Fibrosis Canada, proposing to use H. influenzae's ability to extract H. influenzae DNA from complex mixtures as a tool to characterize H. influenzae populations in respiratory tract samples from children with cystic fibrosis. The post-doc (blessed be his name) wrote this one, with minimal input from me.
A draft of a third proposal, to NSERC (Natural Sciences and Engineering Research Council, Canada's NSF) has been passed on to UBC's internal-review system. It proposes to test the hypothesis that the self-biased uptake systems of the Pasteurellaceae and Neisseria species are due to mechanistic biases in the uptake process rather than to selection for optimal recombination, by looking for similar biases in bacteria that don't preferentially take up their own DNA. The final proposal doesn't need to be submitted until the end of the month.
So I might have time to do an experiment or two. I'll have to ask the Research Associate what I should do.
Anyway, to start off easy, here's today's weather forecast:

Two grant proposals have been submitted. You can get the CIHR one on the 'What we're planning' page of our website (link in the left sidebar); the CFC one will appear there soon. The first is a big one to CIHR (Canadian Institutes for Health Research, Canada's NIH), proposing to develop the information base and algorithm needed to predict transformational recombination in the respiratory tract. The second is a smaller one to Cystic Fibrosis Canada, proposing to use H. influenzae's ability to extract H. influenzae DNA from complex mixtures as a tool to characterize H. influenzae populations in respiratory tract samples from children with cystic fibrosis. The post-doc (blessed be his name) wrote this one, with minimal input from me.
A draft of a third proposal, to NSERC (Natural Sciences and Engineering Research Council, Canada's NSF) has been passed on to UBC's internal-review system. It proposes to test the hypothesis that the self-biased uptake systems of the Pasteurellaceae and Neisseria species are due to mechanistic biases in the uptake process rather than to selection for optimal recombination, by looking for similar biases in bacteria that don't preferentially take up their own DNA. The final proposal doesn't need to be submitted until the end of the month.
So I might have time to do an experiment or two. I'll have to ask the Research Associate what I should do.
Long time no posting. Sorry. I've been working on our new CIHR grant proposal, but for unknown reasons not doing this on the blog.
But I have a point I need to make in the proposal, and to a colleague who's just generously critiqued a draft for us. I'm having a hard time finding a way to explain what I mean, so, of course, I'll try doing it here.

As you can see from the Specific Aims above, our goal isn't just to investigate the factors affecting transformation, but to incorporate these factors into a predictive model (initially two separate sub-models), and to test this model's predictions against the real transformation seen in a laboratory version of the respiratory tract environment. Our hope is that this model or its more sophisticated successors can be used to predict clinically important genetic exchange, leading to modifications in drug and vaccine design that minimize the opportunities for specific exchange events.
Proposals typically describe how the results could be used to improve medical treatments, but for basic science this is usually just a 'somebody, someday' hope. Because the researchers won't themselves be applying their results, there's no incentive to make sure they're in a useable form. For genetic exchange the outcome has been that, although we have lots of descriptive information about the mechanisms and regulation, and lots of surveys of its significance in natural populations, none of the information can be combined into useful predictions.
We however propose to integrate data collection, prediction development, and prediction testing into one coordinated research program. This means that the data we generate about uptake and recombination biases has to be in a form that can be incorporated into computer programs that predict uptake and recombination from input genome sequences. And it means that the predictions of these programs will be tested against reality
But I have a point I need to make in the proposal, and to a colleague who's just generously critiqued a draft for us. I'm having a hard time finding a way to explain what I mean, so, of course, I'll try doing it here.

As you can see from the Specific Aims above, our goal isn't just to investigate the factors affecting transformation, but to incorporate these factors into a predictive model (initially two separate sub-models), and to test this model's predictions against the real transformation seen in a laboratory version of the respiratory tract environment. Our hope is that this model or its more sophisticated successors can be used to predict clinically important genetic exchange, leading to modifications in drug and vaccine design that minimize the opportunities for specific exchange events.
Proposals typically describe how the results could be used to improve medical treatments, but for basic science this is usually just a 'somebody, someday' hope. Because the researchers won't themselves be applying their results, there's no incentive to make sure they're in a useable form. For genetic exchange the outcome has been that, although we have lots of descriptive information about the mechanisms and regulation, and lots of surveys of its significance in natural populations, none of the information can be combined into useful predictions.
We however propose to integrate data collection, prediction development, and prediction testing into one coordinated research program. This means that the data we generate about uptake and recombination biases has to be in a form that can be incorporated into computer programs that predict uptake and recombination from input genome sequences. And it means that the predictions of these programs will be tested against reality
Typical journal spam...
Dear Dr. Rosemary,
Greetings from Journal of Fungal Genomics & Biology.
Based on your eminent contribution in the field of fungal genomics and biology, we are glad to invite you to submit Research/Review article for our prestigious Journal, Fungal Genomics & Biology. Your contribution is of great importance for us and it will help our journal to establish its high standards.
You can submit manuscript at http://www.omicsonline.org/submission/
(OR) by e-mail to editor.fgb@group.org.
You can submit your valuable articles by mid of September, 2012 so that we can publish in the coming issue (or) as per your interest at your earliest convenience.
Please do not hesitate to contact us for any queries.
Awaiting for your positive response.
With Regards,
XXXXXX (name redacted to protect the guilty)
Editorial office
OMICS GROUP INCORPORATION
And no, I've never published ANYTHING on fungal biology.
I tried to get an image of the journal cover, but all the Omics Group page links are broken:
I tried to get an image of the journal cover, but all the Omics Group page links are broken:

Should women ask for more, or are we punished for being 'greedy'?
A short paper in the Lancet (Bedi et al) compares the sizes of grants awarded to women and to men by the Wellcome Trust, from 2000 to 2008. Male applicants get, on average, £44,735 more than women. Women's grants must also be shorter, because the disparity is even more dramatic when funding is calculated per year.

They recommend that mentors 'should ensure that women are as ambitious as men in their outlook, and in their grant proposals'. But they don't consider the alternative explanation, that women who ask for as much as men do are seen as greedy and undeserving, while women with modest ambitions are rewarded. This might be checked by comparing the the requested amount with the probability of being funded, for men and for women.
Their second recommendation, that 'men should be encouraged to be economical when costing such applications', is of course absurd.

Because women and men have similar success rates for these grants, and the amounts awarded are usually the amounts requested, the authors think the discrepancy is because 'women are systematically less ambitious in the amounts of funding requested in their grant applications.'
They recommend that mentors 'should ensure that women are as ambitious as men in their outlook, and in their grant proposals'. But they don't consider the alternative explanation, that women who ask for as much as men do are seen as greedy and undeserving, while women with modest ambitions are rewarded. This might be checked by comparing the the requested amount with the probability of being funded, for men and for women.
Their second recommendation, that 'men should be encouraged to be economical when costing such applications', is of course absurd.
The model that's the goal of our CIHR proposal
In our new CIHR proposal we're going to propose to gather the experimental information needed to make a predictive model of genetic exchange by transformation in the respiratory tract. I've been trying to lay out the Research Plan by describing the things we want to find out. But I realized last night that it might be better to start by describing the predictive model we want to generate, and let its requirements determine what we propose to find out.
That was when I also realized that I have no idea what form the model should take! That is, what would be the input information, and what would be the output. Let's see if I can do better this morning.

Of course, we don't need to have just one model. Several different models might be appropriate, to address different questions. For example, given two H. influenzae genome sequences, what are the relative probabilities of different recombination events? Of course we couldn't evaluate ALL the possible events, but we could tag the most likely and least likely. So we'd want to be able to scan the genome sequences of potential donors and flag the segments most likely and least likely to be taken up by competent cells. And we'd want to scan the genome sequences of potential recipients, flagging the segments most likely and least likely to undergo homologous recombination.
We could do this a different way. For each potential donor genome, we would evaluate the probability that a given segment would be taken up as a DNA fragment by competent cells. Then, for the other genome, we'd calculate the probabilities that different parts of this fragment would be recombined into the recipient genome.
Or we could evaluate a pool of DNA sequences with different proportions from different strains or species, again tagging the segments most likely to be taken up, and the effects of changing the proportions from different sources. And we could consider a population of possible recipients, consisting of strains with different levels of competence and present in different proportions.
OK, I think the post-doc and I are going to spend some time today working on this at the whiteboards in the hall.
That was when I also realized that I have no idea what form the model should take! That is, what would be the input information, and what would be the output. Let's see if I can do better this morning.

Of course, we don't need to have just one model. Several different models might be appropriate, to address different questions. For example, given two H. influenzae genome sequences, what are the relative probabilities of different recombination events? Of course we couldn't evaluate ALL the possible events, but we could tag the most likely and least likely. So we'd want to be able to scan the genome sequences of potential donors and flag the segments most likely and least likely to be taken up by competent cells. And we'd want to scan the genome sequences of potential recipients, flagging the segments most likely and least likely to undergo homologous recombination.
We could do this a different way. For each potential donor genome, we would evaluate the probability that a given segment would be taken up as a DNA fragment by competent cells. Then, for the other genome, we'd calculate the probabilities that different parts of this fragment would be recombined into the recipient genome.
Or we could evaluate a pool of DNA sequences with different proportions from different strains or species, again tagging the segments most likely to be taken up, and the effects of changing the proportions from different sources. And we could consider a population of possible recipients, consisting of strains with different levels of competence and present in different proportions.
OK, I think the post-doc and I are going to spend some time today working on this at the whiteboards in the hall.
A slick puff piece on my research for only £1960!

I just got an email from someone named Simon Jones, offering to publish an article about my research in a publication called International Innovations. Here's their website. They appear to be an online publisher of articles about advances in health care research. Oops, no that was just their health care branch. They have other branches, for Energy, Environment, Nanomaterials etc. You can see the whole list here. They claim to be 'The Leading Global Dissemination Resource'. (Red text is my highlighting.)

They describe what they publish as 'open access journals', but they're certainly not peer reviewed. Instead they're collections of puff pieces, intended to make the researcher look good.
The first part of the email was followed by a long collection of lists, of researchers they have written about, topics they've covered, institutions and countries that subscribe, etc. Far too long to include here. And after that comes the details:

So the deal is: For a fee of £1960 (about $3000) they prepare a 3-page glossy article about how great my research is, based on 2-3 hr of input from me. They apparently do the writing and prepare the figures, and they publish it in their slick online magazine, and perhaps in a glossy paper edition too. I own the rights, so I can then splash this article around in any way I like - send it to my Dean, post it on my web page, print a glossy brochure to hand out...
Given the copyright promise, it's a bit surprising to see this statement in each issue:
Copyright reproduction in whole or part by any means without written permission of the publisher is strictly forbidden. The publisher accepts no responsibility for errors, omissions, or the consequences thereof. The opinions expressed in International Innovation are those of the individual and not endorsed by Research Media. Copyright Research Media.It's also odd that they claim copyright but disclaim any responsibility for the content.
And maybe it's not surprising that the published material nowhere indicates that the researchers being profiled paid for their profiles.
And an application to Cystic Fibrosis Canada
(I wrote this yesterday but forgot to click 'Publish'.)
I've been spending an inordinate amount of time preparing the 'Notification of Intent to Apply for a Discovery Grant' for NSERC, and I submitted that this morning. I was about to get back to writing the CIHR proposal, but then I remembered that Cystic Fibrosis Canada also makes research grants, and since the CIHR proposal is so strongly focused on respiratory infections I thought I should check these out.
Well, they make about 20 grants per year (about 30% of these are new, success rate for new applications about 25%). The application deadline is Oct. 3, which is 2 weeks after the CIHR deadline and a month before the NSERC deadline, so that's feasible, especially as what I'd send them would be a modified (reduced?) version of the CIHR proposal.
They also have an Intent to Apply notification process, and I'd need to email the form tomorrow, August 1. Our chances probably aren't great, as the proposal isn't close to therapy, but it's worth a try.
I've been spending an inordinate amount of time preparing the 'Notification of Intent to Apply for a Discovery Grant' for NSERC, and I submitted that this morning. I was about to get back to writing the CIHR proposal, but then I remembered that Cystic Fibrosis Canada also makes research grants, and since the CIHR proposal is so strongly focused on respiratory infections I thought I should check these out.
Well, they make about 20 grants per year (about 30% of these are new, success rate for new applications about 25%). The application deadline is Oct. 3, which is 2 weeks after the CIHR deadline and a month before the NSERC deadline, so that's feasible, especially as what I'd send them would be a modified (reduced?) version of the CIHR proposal.
They also have an Intent to Apply notification process, and I'd need to email the form tomorrow, August 1. Our chances probably aren't great, as the proposal isn't close to therapy, but it's worth a try.
More on the NSERC proposal
Title: I want something catchy but not frivolous. 'Do bacteria have sex?' has to be immediately followed by one particular definition of sex, as any process evolved (and maintained) by selection for the random genetic recombination it causes. But 'Do bacteria have sex for sex?' might get the point across even in the absence of accompanying definitions of 'sex' and 'sex'. It even communicates the point that there are two evolutionary definitions of sex that could apply to bacteria.
Rationale: I explained this in a post a few days ago:
Outcomes: Here are the three possibilities:

1. Finding such biases in one or more bacteria whose genomes are not enriched for the favoured motif would be strong evidence that the bias exists for mechanistic reasons, not because uptake of self DNA enhances recombination. In principle we only need to find one such case.
2. Finding that DNA uptake has absolutely no sequence biases in one or more of the tested bacteria would greatly weaken our hypothesis. We will have argued that DNA uptake is expected to have sequence biases because the high forces that have been measured on DNA require very strong contacts between DNA and the proteins of the uptake machinery. Such strong contacts always show some sequence-dependence. Finding that uptake is completely unbiased in any species would mean that this argument is invalid.
3. But what if we find that uptake is always biased and the corresponding genomes are always enriched for the preferred motifs? This wouldn't disprove our hypothesis that the bias exists for mechanistic reasons - in fact it's exactly what our model predicts, given that the bacteria we're testing are known to recombine some of the DNA they take up in the lab. But I need to think through the implications more carefully. In the previous post I wrote:
So, if we find accumulation of a preferred motif in bacteria previously thought to have unbiased uptake, we'll know that, in nature as well as in the lab, that the cells must sometimes take up DNA that replaces chromosomal sequences by homologous recombination. So we'll have generalized the bias-plus-abundance phenomenon already characterized in the Neisserias and Pasteurellaceae. This would certainly be seen as an important (publishable) result.
But this wouldn't mean that the recombination this promotes must be beneficial, because drive can explain this accumulation. Would there be any way to tell whether the accumulation has been significantly affected by selection? Are thee tests we could do in other bacteria that we can't do in H. influenzae? Or that we should do in H. influenzae? (Of course we can never rule out that the benefits of recombination are too small to detect...)
If we started by assuming there's no selection for or against the genetic consequences of recombination, we might think we could use the degree of enrichment to infer, using our model, how much recombination must be happening. If (letting go of our assumption) the enrichment was really partly due to selection for recombination, then the actual level of recombination in the population would have to be less than the model predicts. (Yikes, this logic is getting weird.) But I don't think our model is nearly that realistic, and we couldn't use it to make predictions anyway because we don't have enough real information about the properties of any real population.
Enough for now...
Rationale: I explained this in a post a few days ago:
Natural competence, the ability to take up DNA fragments from the environment, is the only bacterial 'parasexual' process for which reasonable doubt remains about its non-recombinational function. This doubt arises from the apparent self-specificity of DNA uptake by bacteria in two groups, the genus Neisseria and the family Pasteurellaceae. Bacteria in these groups preferentially take up DNAs containing short sequence motifs that are ~100-fold more abundant in their own genomes than expected by chance. This match between the bias of the DNA uptake machinery and the genomic abundance of a DNA motif has been interpreted as an adaptation that enhances the presumed recombinational (sexual) benefits of DNA uptake, by allowing mate-choice or excluding possibly harmful foreign DNAs. However a simpler non-sexual explanation exists - that the preferred sequences play purely mechanistic roles in DNA uptake, and that the motifs' abundances in the respective genomes are due to a passive accumulation caused by biased uptake and subsequent unselected recombination.Aims: So the main aim of this proposal would be to test the DNA uptake systems of other naturally competent bacteria for sequence biases.
Outcomes: Here are the three possibilities:

1. Finding such biases in one or more bacteria whose genomes are not enriched for the favoured motif would be strong evidence that the bias exists for mechanistic reasons, not because uptake of self DNA enhances recombination. In principle we only need to find one such case.
2. Finding that DNA uptake has absolutely no sequence biases in one or more of the tested bacteria would greatly weaken our hypothesis. We will have argued that DNA uptake is expected to have sequence biases because the high forces that have been measured on DNA require very strong contacts between DNA and the proteins of the uptake machinery. Such strong contacts always show some sequence-dependence. Finding that uptake is completely unbiased in any species would mean that this argument is invalid.
3. But what if we find that uptake is always biased and the corresponding genomes are always enriched for the preferred motifs? This wouldn't disprove our hypothesis that the bias exists for mechanistic reasons - in fact it's exactly what our model predicts, given that the bacteria we're testing are known to recombine some of the DNA they take up in the lab. But I need to think through the implications more carefully. In the previous post I wrote:
If we do find overrepresentation, we can (1) decide whether this overrepresentation would create any significant degree of preferential uptake of self-DNA, and (2) use our simulation model (or refinements of it) to evaluate how much recombination must be going on to give this overrepresentation.The molecular drive that happens in the simulation model is the null hypothesis for uptake sequences; it shows how sequences preferred by the uptake machinery accumulate in the genome, without invoking any benefit from recombination. If uptake has a sequence bias, the DNA taken up is sometimes homologous to the cell's genome, and this DNA sometimes recombines with the chromosome, we expect the preferred sequences to slowly accumulate in the chromosome. If recombination of chromosomal alleles has a net genetic benefit, selection for the self-uptake promoted by uptake sequences will act in addition to drive (not instead of it). If recombination instead has a net cost, selection against it may oppose drive and reduce accumulation of the sequences preferred by the uptake machinery.
So, if we find accumulation of a preferred motif in bacteria previously thought to have unbiased uptake, we'll know that, in nature as well as in the lab, that the cells must sometimes take up DNA that replaces chromosomal sequences by homologous recombination. So we'll have generalized the bias-plus-abundance phenomenon already characterized in the Neisserias and Pasteurellaceae. This would certainly be seen as an important (publishable) result.
But this wouldn't mean that the recombination this promotes must be beneficial, because drive can explain this accumulation. Would there be any way to tell whether the accumulation has been significantly affected by selection? Are thee tests we could do in other bacteria that we can't do in H. influenzae? Or that we should do in H. influenzae? (Of course we can never rule out that the benefits of recombination are too small to detect...)
If we started by assuming there's no selection for or against the genetic consequences of recombination, we might think we could use the degree of enrichment to infer, using our model, how much recombination must be happening. If (letting go of our assumption) the enrichment was really partly due to selection for recombination, then the actual level of recombination in the population would have to be less than the model predicts. (Yikes, this logic is getting weird.) But I don't think our model is nearly that realistic, and we couldn't use it to make predictions anyway because we don't have enough real information about the properties of any real population.
Enough for now...
NSERC Proposal
Yikes, even though our proposal to NSERC (I think that stands for the Natural Sciences and Engineering Research Council) wouldn't be due until November 1, I just discovered that we need to get a 'Notification of Intent' in to them by next Wednesday (August 1). It hadn't occurred to me that NSERC had an advance registration requirement, and, if it did, I would have expected registration to be due a month before the grant submission deadline, as is the case for CIHR.
Luckily the people who send UBC faculty 'Grant Opportunity Updates' thought to include a reminder about this - otherwise I'd have missed the registration deadline for sure. If they're like CIHR, there's no recourse for missing the deadline, but it's worse for NSERC because they have only one submission deadline each year.
And of course NSERC doesn't seem to use the 'Common CV' system that most other agencies do, so I have to input all my information again...
And, because NSERC does send proposals out to external reviewers, unlike CIHR, I need to come up with a list of 5 possible reviewers. That will take some thinking. While I'm at it, I'll also ask for pointers from friends who regularly apply to NSERC.
Luckily the people who send UBC faculty 'Grant Opportunity Updates' thought to include a reminder about this - otherwise I'd have missed the registration deadline for sure. If they're like CIHR, there's no recourse for missing the deadline, but it's worse for NSERC because they have only one submission deadline each year.
And of course NSERC doesn't seem to use the 'Common CV' system that most other agencies do, so I have to input all my information again...
And, because NSERC does send proposals out to external reviewers, unlike CIHR, I need to come up with a list of 5 possible reviewers. That will take some thinking. While I'm at it, I'll also ask for pointers from friends who regularly apply to NSERC.
Are proteins wet and gooey?

To me this protein image looks wet and gooey. Initially I found that disconcerting, because I'm used to the dry solid appearance of the more standard visualizations. But now I think that 'wet and gooey' is actually a realistic description of the surface properties of any protein.
And a possible NSERC proposal
If we decide to follow the CIHR proposal outline I've made in the previous post, we'll probably do a separate proposal to NSERC on the role of uptake sequences in the mechanism of DNA uptake. I was just discussing this with the post-doc and he had a great idea about also using the analysis to make inferences about the frequencies of chromosomal recombination in naturally competent species. So I'm going to write it out here quickly, before I forget it.
 Motivation: This work addresses a very important question with big/deep/fundamental importance to the colossal problem of the origin of meiotic sex in eukaryotes. The question is 'Do bacteria have any processes that evolved because of selection for recombination of chromosomal alleles?' We think this selection is the reason for the success of meiotic sexual reproduction in eukaryotes, but compelling evidence for this has been elusive. Bacteria have four well-studied processes that do generate homologous recombination; three that transfer DNA between cells and one that carries out homologous recombination. But almost every aspect of these processes has been shown to cause recombination as an unselected side effect of processes selected for other functions.
Motivation: This work addresses a very important question with big/deep/fundamental importance to the colossal problem of the origin of meiotic sex in eukaryotes. The question is 'Do bacteria have any processes that evolved because of selection for recombination of chromosomal alleles?' We think this selection is the reason for the success of meiotic sexual reproduction in eukaryotes, but compelling evidence for this has been elusive. Bacteria have four well-studied processes that do generate homologous recombination; three that transfer DNA between cells and one that carries out homologous recombination. But almost every aspect of these processes has been shown to cause recombination as an unselected side effect of processes selected for other functions.
Natural competence, the ability to take up DNA fragments from the environment, is the only one of the four for which reasonable doubt remains about its non-recombinational function. The strongest selection for DNA uptake is generated by its nutritional consequences; DNA is an excellent and economical source of preformed nucleotides and of phosphate, and the nutrient function of DNA uptake is supported by its regulation by nutritional signals in many bacteria. This doubt arises from the apparent self-specificity of DNA uptake by bacteria in two groups, the genus Neisseria and the family Pasteurellaceae. Bacteria in these groups preferentially take up DNAs containing short sequence motifs that are ~100-fold more abundant in their own genomes than expected by chance. This match between the bias of the DNA uptake machinery and the genomic abundance of a DNA motif has been interpreted as an adaptation that enhances the presumed recombinational (sexual) benefits of DNA uptake, by allowing mate-choice or excluding possibly harmful foreign DNAs. However a simpler non-sexual explanation exists - that the preferred sequences play purely mechanistic roles in DNA uptake, and that the motifs' abundances in the respective genomes are due to a passive accumulation caused by biased uptake and subsequent unselected recombination. (Something here about why mechanistic sequence biases are plausible/expected.)
Only a subset of competent species exhibit this uptake specificity (strongly biased uptake and strongly enriched genome). The others are considered to have no uptake bias at all (and no genomic uptake sequences), largely because they show no preference for DNA fragments form their own genomes over those from other genomes. Thus the most powerful test of this hypothesis is testing these species for cryptic uptake biases. Finding of even minor biases in their uptake machinery would confirm that biased uptake need not result from selection for mate-choice. We have developed a simple and very powerful method to test for such biases, using Illumina sequencing of DNA fragments taken up from pools of highly degenerate DNA fragments.
As a bonus, this analysis will allow estimation, for a number of bacterial species, of the frequencies of chromosomal recombination between close relatives. We will be working with species whose genomes have been sequenced. Once any uptake biases have been identified, the corresponding genomes can be analyzed to detect any enrichment of the preferred motifs. (Unless the motifs are as complex as the known Neisserial and Pasteurellacean ones) For motifs that are either short or not very strong, finding enrichment will not be evidence for a sexual function, since the same motifs will occur frequently in foreign DNAs. However the enrichment will allow estimation of the frequency of recombination in the species, because we have developed a computer-simulation model of the accumulation process.
Aims:
A. Test naturally competent bacteria for biases in their DNA uptake mechanisms. We've done H. influenzae, which has well characterized uptake bias. We'll test Acinetobacter bayleyi and Thermus thermophilus, both Gram-negative bacteria thought to have no uptake bias and no genomic enrichment. I've already arranged collaborations with a researcher; it will probably be simplest to travel to their lab to do the uptake experiments. We'll use fully degenerate Illumina-ready DNA fragments, which we have already designed and ordered. The analysis methods for the Illumina (or Miseq) output have already been developed for H. influenzae; they may need modification for the fully degenerate sequences. We could also test a Vibrio (maybe not cholerae!) and maybe Pseudomonas stutzeri. We could also test bacteria that have some uptake bias, with (A. pleuropneumoniae and G. anatis) or without (Campylobacter) genomic uptake motifs, but this wouldn't really test our hypothesis.
Could we also test Gram-positive bacteria for uptake bias (Streptococcus or Bacillus)? This is the equivalent of testing Gram-negative bacteria for translocation bias, and will be substantially more difficult because as the DNA enters the cytoplasm it becomes single-stranded and partly degraded (at one end). But we think we can develop ways to fish out the single strands from the cytoplasm.
What if we find no evidence of bias? I'd be surprised. This wouldn't disprove our hypothesis, but it would weaken it, because our hypothesis is built on the expectation that all DNA binding proteins have some sequence specificity, and that tasks requiring application of force to DNA will benefit from the tighter binding created by sequence-specific interactions.
B. Examine the corresponding genome sequences for overrepresentation of the preferred motifs. If we find no overrepresentation, we will have established that uptake biases need not result from selection for preferential uptake of self-DNA. If we do find overrepresentation, we can (1) decide whether this overrepresentation would create any significant degree of preferential uptake of self-DNA, and (2) use our simulation model (or refinements of it) to evaluate how much recombination must be going on to give this overrepresentation.
Back to the big picture: If we find uptake bias without genomic overrepresentation, or without self-specificity, we'll have swept away the last feeble pillar still propping up the claim that bacteria have processes that evolved for genetic exchange.

Natural competence, the ability to take up DNA fragments from the environment, is the only one of the four for which reasonable doubt remains about its non-recombinational function. The strongest selection for DNA uptake is generated by its nutritional consequences; DNA is an excellent and economical source of preformed nucleotides and of phosphate, and the nutrient function of DNA uptake is supported by its regulation by nutritional signals in many bacteria. This doubt arises from the apparent self-specificity of DNA uptake by bacteria in two groups, the genus Neisseria and the family Pasteurellaceae. Bacteria in these groups preferentially take up DNAs containing short sequence motifs that are ~100-fold more abundant in their own genomes than expected by chance. This match between the bias of the DNA uptake machinery and the genomic abundance of a DNA motif has been interpreted as an adaptation that enhances the presumed recombinational (sexual) benefits of DNA uptake, by allowing mate-choice or excluding possibly harmful foreign DNAs. However a simpler non-sexual explanation exists - that the preferred sequences play purely mechanistic roles in DNA uptake, and that the motifs' abundances in the respective genomes are due to a passive accumulation caused by biased uptake and subsequent unselected recombination. (Something here about why mechanistic sequence biases are plausible/expected.)
Only a subset of competent species exhibit this uptake specificity (strongly biased uptake and strongly enriched genome). The others are considered to have no uptake bias at all (and no genomic uptake sequences), largely because they show no preference for DNA fragments form their own genomes over those from other genomes. Thus the most powerful test of this hypothesis is testing these species for cryptic uptake biases. Finding of even minor biases in their uptake machinery would confirm that biased uptake need not result from selection for mate-choice. We have developed a simple and very powerful method to test for such biases, using Illumina sequencing of DNA fragments taken up from pools of highly degenerate DNA fragments.
As a bonus, this analysis will allow estimation, for a number of bacterial species, of the frequencies of chromosomal recombination between close relatives. We will be working with species whose genomes have been sequenced. Once any uptake biases have been identified, the corresponding genomes can be analyzed to detect any enrichment of the preferred motifs. (Unless the motifs are as complex as the known Neisserial and Pasteurellacean ones) For motifs that are either short or not very strong, finding enrichment will not be evidence for a sexual function, since the same motifs will occur frequently in foreign DNAs. However the enrichment will allow estimation of the frequency of recombination in the species, because we have developed a computer-simulation model of the accumulation process.
Aims:
A. Test naturally competent bacteria for biases in their DNA uptake mechanisms. We've done H. influenzae, which has well characterized uptake bias. We'll test Acinetobacter bayleyi and Thermus thermophilus, both Gram-negative bacteria thought to have no uptake bias and no genomic enrichment. I've already arranged collaborations with a researcher; it will probably be simplest to travel to their lab to do the uptake experiments. We'll use fully degenerate Illumina-ready DNA fragments, which we have already designed and ordered. The analysis methods for the Illumina (or Miseq) output have already been developed for H. influenzae; they may need modification for the fully degenerate sequences. We could also test a Vibrio (maybe not cholerae!) and maybe Pseudomonas stutzeri. We could also test bacteria that have some uptake bias, with (A. pleuropneumoniae and G. anatis) or without (Campylobacter) genomic uptake motifs, but this wouldn't really test our hypothesis.
Could we also test Gram-positive bacteria for uptake bias (Streptococcus or Bacillus)? This is the equivalent of testing Gram-negative bacteria for translocation bias, and will be substantially more difficult because as the DNA enters the cytoplasm it becomes single-stranded and partly degraded (at one end). But we think we can develop ways to fish out the single strands from the cytoplasm.
What if we find no evidence of bias? I'd be surprised. This wouldn't disprove our hypothesis, but it would weaken it, because our hypothesis is built on the expectation that all DNA binding proteins have some sequence specificity, and that tasks requiring application of force to DNA will benefit from the tighter binding created by sequence-specific interactions.
B. Examine the corresponding genome sequences for overrepresentation of the preferred motifs. If we find no overrepresentation, we will have established that uptake biases need not result from selection for preferential uptake of self-DNA. If we do find overrepresentation, we can (1) decide whether this overrepresentation would create any significant degree of preferential uptake of self-DNA, and (2) use our simulation model (or refinements of it) to evaluate how much recombination must be going on to give this overrepresentation.
Back to the big picture: If we find uptake bias without genomic overrepresentation, or without self-specificity, we'll have swept away the last feeble pillar still propping up the claim that bacteria have processes that evolved for genetic exchange.
A new CIHR proposal?
The more I think about the 'significance' part of our latest CIHR proposal, the weaker I think it was. So here I'm going to lay out the bones of a different proposal, one that builds very well on our current strengths but allows much stronger claims of significance.
Possible title: Predicting the outcomes of genetic exchange in polyclonal infections
The problem: Genetic exchange between closely related pathogens can increase virulence and is responsible for many failed attempts at control (spread of antibiotic resistance, escape from immune surveillance and vaccine immunity). At present we have no ways to anticipate or prevent this, largely because we are ignorant of the constraints and biases of the underlying steps. Previous (sparse?) attempts to understand genetic exchange have been based on inferences from (i) laboratory studies of the mechanism and regulation of DNA-transfer processes and recombination and (ii) detection of past recombination events in natural populations. The former bear little relation to events in real populations, and the latter are confounded by later time, genetic drift and natural selection.
Hypothesis: Identifying the constraints on transformational genetic exchange will allow the outcomes of natural recombination events to be predicted.
Significance: The ability to predict the most likely genetic exchange events will help researchers prepare for new variant strains of bacteria. Identifying the causes of the constraints may also permit interventions that block genetic exchange in polyclonal infections such as the cystic fibrosis lung.
Aims:
A. Identify the DNA sequence effects that constrain DNA uptake.
B. Identify the constraints on homologous recombination between H. influenzae strains.
C. Use the results of the above studies to develop a probabilistic model of recombination, and test these predictions using datasets of recombination events from natural and simulated bacterial communities.

On another topic, I've been getting emails about a new paper in the Journal of Biological Chemistry, which claims to explain the growth of GFAJ-1 by use of phosphate from degraded ribosomes. I don't think this idea is sound, but I'm going to leave it for Carmen Drahl and Ed Yong to deal with.
Possible title: Predicting the outcomes of genetic exchange in polyclonal infections
The problem: Genetic exchange between closely related pathogens can increase virulence and is responsible for many failed attempts at control (spread of antibiotic resistance, escape from immune surveillance and vaccine immunity). At present we have no ways to anticipate or prevent this, largely because we are ignorant of the constraints and biases of the underlying steps. Previous (sparse?) attempts to understand genetic exchange have been based on inferences from (i) laboratory studies of the mechanism and regulation of DNA-transfer processes and recombination and (ii) detection of past recombination events in natural populations. The former bear little relation to events in real populations, and the latter are confounded by later time, genetic drift and natural selection.
Hypothesis: Identifying the constraints on transformational genetic exchange will allow the outcomes of natural recombination events to be predicted.
Significance: The ability to predict the most likely genetic exchange events will help researchers prepare for new variant strains of bacteria. Identifying the causes of the constraints may also permit interventions that block genetic exchange in polyclonal infections such as the cystic fibrosis lung.
Aims:
A. Identify the DNA sequence effects that constrain DNA uptake.
- Clarify the sequence biases of DNA uptake by H. influenzae.
- Identify the proteins that interact with the preferred sequences.
- Identify sequence biases of DNA uptake by other bacteria, especially those not known to exhibit bias.
- Characterize the effects of these biases in natural and simulated communities.
B. Identify the constraints on homologous recombination between H. influenzae strains.
- Identify the effects of DNA sequences and sequence heterologies on the extents and endpoints of recombination tracts.
- Identify the effects of DNA sequences and sequence heterologies on recombination frequencies across the H. influenzae genome
C. Use the results of the above studies to develop a probabilistic model of recombination, and test these predictions using datasets of recombination events from natural and simulated bacterial communities.
- A dataset of past recombination events inferred from genome sequence data of H. influenzae strains.
- A metagenomic dataset derived from a short-term evolution of a polyclonal H. influenzaelaboratory culture.
- A metagenomic dataset from cystic fibrosis sputum.
The figure shows the structure of H. influenzae recombination tracts (data from Mell et al. Transformation of natural genetic variation into Haemophilus influenzae. PLoS Pathog 7(7): e1002151. doi:10.1371/journal.ppat.1002151)

On another topic, I've been getting emails about a new paper in the Journal of Biological Chemistry, which claims to explain the growth of GFAJ-1 by use of phosphate from degraded ribosomes. I don't think this idea is sound, but I'm going to leave it for Carmen Drahl and Ed Yong to deal with.
Subscribe to:
Posts (Atom)