














I repeated the fraction-competent assay on log-phase murE749 hypercompetent cells, as I said I should in the previous post. I did a very thorough and well-controlled experiment, but the results tell me that this isn't a very reliable measure of how much the cells in the culture differ in their competence.
I grew the cells at low density in rich medium for about 3.5 hours, so they would all be growing exponentially (in log phase). I added MAP7 DNA to the cells, let them grow for 15 minutes, and added DNase I to prevent continuing DNA uptake. I then let the cells continue growing for another 1.5 hours, to allow all the antibiotic resistance alleles to be fully expressed. Then I diluted the culture and spread the cells on agar plates containing different antibiotics, singly or in pairwise combinations.
MAP7 DNA contains point mutations causing resistance to 7 different antibiotics, but I only selected for 4 of them in this experiment: novobiocin (nov), kanamycin (kan), spectinomycin (spc) and nalidixic acid (nal). The nov and kan alleles are close together on the chromosome, so I didn't select for those two together, but I selected for nov+spc, noc+nal, nal+spc, kan+nal, and kan+spc. These combinations gave me 5 different measures of fraction competent.
The nal allele gave a low transformation frequency on its own (4.9x10^-4), and all 3 of the combinations that included nal gave low estimates of fraction competent: 0.06, 0.08 and 0.09. The other two combinations gave higher estimates: 0.28 and 0.58.
That's a ten-fold range of the estimates. Practically, the difference between 0.06 of the cells being competent and 0,58 being competent is enormous, but the fraction competent assays can't tell the difference.
A former student had proposed developing a fluorescent reporter-gene assay that would let us look at cells under the microscope and count the ones that had turned on their competence genes. I still think it would be a big pain, largely because the cells are so small, but maybe the recent improvements in reporter molecules and in microscopy now make this a good idea.
Field of Science
-
-
Change of address1 year ago in Variety of Life
-
Change of address1 year ago in Catalogue of Organisms
-
-
Earth Day: Pogo and our responsibility1 year ago in Doc Madhattan
inconclusive fraction-competent results
A few posts ago I described surprising results from an experiment measuring the fractions of the cells in different cultures that were competent. Here they are again:


Conditions were a bit different this time. First, the KW20 (wildtype) culture had not been induced to maximum competence this time - these cells were approaching stationary phase and are 50-100-times less comepetent. Second, this time I remembered to give all the cultures 60 minutes in rich medium before plating to allow expression fo the spectinomycin resistance.
Including expression time doesn't appear to have significantly changes the transformation frequencies for SpcR selected alone, but it substantially increased the double transformation frequencies (NovR SpcR), and this reduced the apparent fraction competent to below 1.0.
So this new experiment clarifies why I got the anomalously high FC in the previous experiment. Unfortunately it doesn't address the reason I wanted to measure FC in the hypercompetent mutants in the first place. The question arose from the analysis of the ∆HI0659 mutant's growth rates. In this experiment). I had found that cells carrying the HI0659 mutant grew normally.
I did this experiment because I wanted to find out whether unopposed expression of the HI0660 'toxin' harms cells (killing them or inhibiting their growth). In the ∆HI0659 cells the HI0660 'toxin' is induced but not opposed by the HI0659 'antitoxin' when competence genes are on. Because competence genes aren't normally on in growing cells anyway, I had tested growth in two hypercompetent mutant backgrounds, sxy-1 and murE749 - this was normal too.
The fraction competent experiments were intended to test whether most of the cells in the hypercompetent cultures has their competence genes on - if not then we might not see a dramatic growth difference even if the toxin does harm cells. But I foolishly did them with cells approaching stationary phase, when I should have done them with cells in exponential growth. I don't need to bother doing this for the sxy-1 mutant, since I already know that only a small fraction of its cells are competent then (I did this experiment years ago), but I should test it for murE749.

And here are the new results:

Including expression time doesn't appear to have significantly changes the transformation frequencies for SpcR selected alone, but it substantially increased the double transformation frequencies (NovR SpcR), and this reduced the apparent fraction competent to below 1.0.
So this new experiment clarifies why I got the anomalously high FC in the previous experiment. Unfortunately it doesn't address the reason I wanted to measure FC in the hypercompetent mutants in the first place. The question arose from the analysis of the ∆HI0659 mutant's growth rates. In this experiment). I had found that cells carrying the HI0659 mutant grew normally.
I did this experiment because I wanted to find out whether unopposed expression of the HI0660 'toxin' harms cells (killing them or inhibiting their growth). In the ∆HI0659 cells the HI0660 'toxin' is induced but not opposed by the HI0659 'antitoxin' when competence genes are on. Because competence genes aren't normally on in growing cells anyway, I had tested growth in two hypercompetent mutant backgrounds, sxy-1 and murE749 - this was normal too.
The fraction competent experiments were intended to test whether most of the cells in the hypercompetent cultures has their competence genes on - if not then we might not see a dramatic growth difference even if the toxin does harm cells. But I foolishly did them with cells approaching stationary phase, when I should have done them with cells in exponential growth. I don't need to bother doing this for the sxy-1 mutant, since I already know that only a small fraction of its cells are competent then (I did this experiment years ago), but I should test it for murE749.
hfq knockout results
I've now examined the effects of knocking out the small RNA-regulating protein Hfq under a wide range of conditions, testing our hypothesis that it regulates competence by helping unfold the translation-inhibiting stem of sxy mRNA.
In my previous experiment I found that the hfq knockout (∆hfq) causes a ten-fold decrease in transformation, both during growth in rich medium and after transfer to the starvation medium MIV. This time I also tested the mutation in combination with either of two hypercompetence-causing mutations (sxy-1 and murE749), and under culture conditions. The reasoning was that if ∆hfq's transformation defect is due to a defect in sxy translation, it should be reduced or eliminated by the sxy-1 mutation, which we know destabilizes the RNA stem. Seeing a similar effect of the murE749 mutation might suggest that this mutation also acts by destabilizing an RNA pairing structure, perhaps the same sxy mRNA stem.
Here are the results.

Starting from the bottom up: In the competence-inducing medium MIV we see the same ~10-fold defect in the wildtype background but no defect in the sxy-1 or murE749 backgrounds. This supports the above hypothesis and suggests that the murE749 mutation also acts by disrupting RNA pairing.
We think that transfer to MIV medium causes two events that together cause expression of the competence genes: (i) cAMP levels go up, and (ii) the mRNA stem no longer blocks translation of sxy mRNA into Sxy protein. Simply adding cAMP to log-phase cells induces only a low level of competence, since the mRNA stem continues to block its translation. This predicts that adding cAMP to hfq mutants will give 10-fold lower competence, but instead we see that competence is nearly normal in the wildtype background and fully normal in the sxy-1 or murE749 backgrounds. This suggests that ∆hfq's competence defect is not duo to a defect in destabilizing the sxy mRNA stem, but instead to an effect on intracellular cAMP levels.
In late-log cells (in rich medium) we think that the low-level competence normally observed is due to a spontaneous increase in cAMP levels, not to destabilization of the sxy mRNA stem. But the experiment saw a larger-than-expected defect in the wildtype background, a ~10-fold defect in the sxy-1 background, and no defect in the murE749 background. I don't know what to make of this.
The final condition was 'overnight cultures' - cultures that grew to maximum density and remained at 37°C on the roller wheel until morning. The hfq+ and ∆hfq cultures in the wildtype background gave no transformants at all, but both hypercompetent backgrounds showed much stronger competence defects than under other conditions (>100-fold). However this could be an artefact of the cessation of growth on expression of the novobiocin resistance allele.
Overall, what should we conclude? I find the cAMP results to be the most compelling; they strongly suggest that our hypothesis is wrong; Hfq does not contribute to the translatability of sxy mRNA.
In my previous experiment I found that the hfq knockout (∆hfq) causes a ten-fold decrease in transformation, both during growth in rich medium and after transfer to the starvation medium MIV. This time I also tested the mutation in combination with either of two hypercompetence-causing mutations (sxy-1 and murE749), and under culture conditions. The reasoning was that if ∆hfq's transformation defect is due to a defect in sxy translation, it should be reduced or eliminated by the sxy-1 mutation, which we know destabilizes the RNA stem. Seeing a similar effect of the murE749 mutation might suggest that this mutation also acts by destabilizing an RNA pairing structure, perhaps the same sxy mRNA stem.
Here are the results.

We think that transfer to MIV medium causes two events that together cause expression of the competence genes: (i) cAMP levels go up, and (ii) the mRNA stem no longer blocks translation of sxy mRNA into Sxy protein. Simply adding cAMP to log-phase cells induces only a low level of competence, since the mRNA stem continues to block its translation. This predicts that adding cAMP to hfq mutants will give 10-fold lower competence, but instead we see that competence is nearly normal in the wildtype background and fully normal in the sxy-1 or murE749 backgrounds. This suggests that ∆hfq's competence defect is not duo to a defect in destabilizing the sxy mRNA stem, but instead to an effect on intracellular cAMP levels.
In late-log cells (in rich medium) we think that the low-level competence normally observed is due to a spontaneous increase in cAMP levels, not to destabilization of the sxy mRNA stem. But the experiment saw a larger-than-expected defect in the wildtype background, a ~10-fold defect in the sxy-1 background, and no defect in the murE749 background. I don't know what to make of this.
The final condition was 'overnight cultures' - cultures that grew to maximum density and remained at 37°C on the roller wheel until morning. The hfq+ and ∆hfq cultures in the wildtype background gave no transformants at all, but both hypercompetent backgrounds showed much stronger competence defects than under other conditions (>100-fold). However this could be an artefact of the cessation of growth on expression of the novobiocin resistance allele.
Overall, what should we conclude? I find the cAMP results to be the most compelling; they strongly suggest that our hypothesis is wrong; Hfq does not contribute to the translatability of sxy mRNA.
Choosing a journal for your manuscript
Listening to Bruce Dancik's #CSPPubTour12 talk yesterday about choosing a journal and submitting your manuscript got me thinking about issues he didn't emphasize. I started with a few, but my list keeps getting longer and longer:
Access to your article: Does the journal provide immediate open access for all its papers? Is this an option, for an extra charge? Open
access after 6 months or a year? Subscription
only? If subscription access, how widely
is it subscribed to?
Cost of publishing: Are there page charges? Optional or required publication charges for open access? Charges for colour figures (only an issue for print journals)?
Limits on article length: No limit? Very
tight? Charges for extra pages?
Likelihood of acceptance: Do your subject, approach and results fit the mandate of the
journal (is yours the kind of manuscript they’re looking for)?
Prestige for your CV: How good is the journal's reputation? How high is its impact factor?
Prestige for journalists: Are papers from this journal often reported in the mainstream media?
Readership:
Does the journal cover a specialized topic or a broad area of science? Which kind of audience are you writing for?
Ease of finding for readers: Is the journal indexed by everything? How well can you use keywords in the
title and abstract to bring in readers from Google Scholar and other search
engines?
Cost of publishing: Are there page charges? Optional or required publication charges for open access? Charges for colour figures (only an issue for print journals)?
Online supplementary materials: Does the journal host these? What are the limitations?
Copyright and licensing: Must you sign away your rights? Can others reuse your material (e.g. use your figures in teaching)?
Turnaround time:
Rapid pre-screening? Total
time from submission to publication?
Online early access?
Is this an ethical publisher? Elsevier? Other
for-profit? Society journal? Predatory publisher (see Beall's list)?
Type of publication:
Online-only? Print edition
only? Both?
Tiresomeness of Instructions to Authors: Will getting your figures into the obscure required format force you to spend $500 on the full version of Photoshop?
Might the journal highlight your paper? Does it include a News and Views or other section that highlights some papers in each issue?
Relative activities of comM from strains Rd and NP
A month or so ago I described four experiments I wanted to do. I've now done the last of them, testing whether the comM gene of strain NP could be partly responsible for that strain's 100-fold lower transformability.
...and the answer appears to be... NO (with one qualification).
The experiment was to compare the abilities of plasmid-borne Rd and NP comM genes to restore full competence to Rd cells whose chromosomal comM gene had been deleted. The RA made all the strains for me - all I had to do was measure their transformation frequencies after fully inducing competence by transfer to starvation medium. The first four bars in the graph below show the results:

Both comM alleles restore normal competence to the Rd knockout when cloned in the forward orientation but not when cloned in the reverse orientation. I think each insert has its own CRP-S promoter, but the plasmid also carries the E. coli lacZ promoter which, I suspect, interferes with expression of inserts whose promoters face in the reverse orientation.
The motivation behind this experiment was a recombinant strain identified by an undergraduate who was working with the postdoc. This strain transforms only 10% as well as Rd; it has been sequenced and we know it contains a single 40 kb segment of NP sequence. The only known competence gene in this 40 kb segment is comM, so they hypothesized that a partially defective NP comM might be responsible for the recombinant's 10-fold reduced competence and partly responsible for NP's 100-fold lower competence. This new result suggests that this hypothesis is wrong.
The qualification is that strong expression from a plasmid could mask lower expression or catalytic activity of NP's comM gene. The gold standard experiment would be to replace the Rd chromosomal allele with the NP version and vice versa. For various technical reasons we haven't been able to do this yet.
...and the answer appears to be... NO (with one qualification).
The experiment was to compare the abilities of plasmid-borne Rd and NP comM genes to restore full competence to Rd cells whose chromosomal comM gene had been deleted. The RA made all the strains for me - all I had to do was measure their transformation frequencies after fully inducing competence by transfer to starvation medium. The first four bars in the graph below show the results:

The motivation behind this experiment was a recombinant strain identified by an undergraduate who was working with the postdoc. This strain transforms only 10% as well as Rd; it has been sequenced and we know it contains a single 40 kb segment of NP sequence. The only known competence gene in this 40 kb segment is comM, so they hypothesized that a partially defective NP comM might be responsible for the recombinant's 10-fold reduced competence and partly responsible for NP's 100-fold lower competence. This new result suggests that this hypothesis is wrong.
The qualification is that strong expression from a plasmid could mask lower expression or catalytic activity of NP's comM gene. The gold standard experiment would be to replace the Rd chromosomal allele with the NP version and vice versa. For various technical reasons we haven't been able to do this yet.
A rotation student replaced the recombinants NP comM allele with the knocked-out Rd version - this reduced its transformation frequency by another 10-fold, about the same as the TF of a simple RD ∆comM strain.
I had tried to transform a ∆comM strain of NP with the Rd comM allele, but the ∆comM mutant the RA made was, unexpectedly, completely non-transformable. She has now given me another NP ∆comM isolate to test. That's the last column in the chart; the red star indicates no transformant colonies at all, so this isolate too is completely non-transformable. This result is consistent with the similarity of the Rd and NP comM plasmid results; they both suggest that the NP comM gene is fully functional, and that some other difference(s) must be responsible for its lower transformability.
So why does the recombinant have lower transformability? Before designing any more experiments I need to be better able to think about the relative chromosomal locations of the various selectable markers and competence genes we're interested in. To this end I'm having our work-study student take a break from glassware-washing and media preparation to do what she calls an 'arts and crafts' project - making us a poster showing the locations of all these genes drawn on a circular chromosome. I just need to give her a list of the genes/ locations to include on her poster.
He takes after me!
Here's a photo of my nephew's science project. He represented the features of the eukaryote cell using cake and candy. The nucleus is a peanut-butter cup, with a jaw-breaker nucleolus. He got a mark of 100%.


A new twist on the fraction-competent problem
On Sunday I attempted to measure what fraction of the cells in some cultures were competent (able to take up DNA fragments and recombine them into their chromosome). I've previously written about this kind of analysis here and here.
Usually when I do this kind of experiment I find that only some cells were competent (anywhere from about 10% to about 50%). But this time I got a very different result, and I don't know why.
I was testing three strains - wildtype cells (strain KW20) and two hypercompetent mutant derivatives, carrying the sxy1 and murE749 mutations. Both KW20 and sxy1 have been tested several times before; mutrE749 hasn't.
The procedure is simple. Transform cells with MAP7 chromosomal DNA and select for two antibiotic resistance mutations located far apart on the chromosome (so they won't ever be carried on the same DNA fragment). Select for each resistance separately and for the two together (double-transformants). Count the resulting colonies and calculate the transformation frequency for each resistance separately and for the double-transformants.
Calculate the fraction competent as the product of the two single-mutation transformation frequencies divided by the double-transformant frequency and by a fudge factor somewhere between 2 and 4.
The fudge factor incorporates two sub-factors accounting for different effects. The first effect is the chance that a single cell took up and recombined two DNA fragments, each containing one of the mutations, but that the incoming DNAs recombined with different strands so that, when the cell divided, one daughter cell got one mutation and the other got the other. This factor is complicated by the unknown effects of mismatch repair, but on average 2 is the appropriate value. The second effect is whether the cells did any cell divisions before they were placed on the agar to grow into colonies. If they did, then cells transformed by a single mutation will have given rise to one resistant colony and one sensitive colony. This factor should have a value of 2 if all the cells had time to divide before plating (if, for example, they needed time to express the antibiotic resistance), by less than 2 if only some did, and by 1 if the cells were plated immediately after transformation. In my experiment I didn't allow any expression time* so this second factor should be closer to 1 than 2. For simplicity I'll use a complete fudge factor of 3.
Here are the calculations:
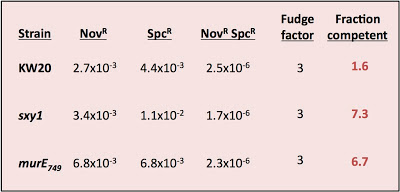
What's wrong with these nice numbers? The fraction competent should ALWAYS be less than 1; that's why it's called a fraction!
Might my assumptions be invalid? This analysis requires that cells be able to take up more than one fragment of DNA, and that taking up one fragment does not affect the probability of taking up another fragment. Because previous experiments have always given values less than 1, we've been assuming that this requirement is met. But this new result only makes sense if many of the cells could only take up one fragment of DNA.
Might my DNA or plates be faulty? The problem isn't due to using some new DNA prep with different properties; I used a fresh tube of the same MAP7 DNA stock we've been using for years. Could it be because I forgot to give the cells some expression time after DNA uptake? Might the lack of expression time reduced the numbers of double transformants (SpcR NovR) much more than it reduced the numbers of single SpcR transformants? But lack of expression time doesn't appear to have been a problem, since I got at least as many SpcR transformants as NovR transformants. The DNase I stock could be a problem. The transformation reactions are stopped after 15 minutes by adding DNase I to degrade the remaining DNA - a few days ago I tested the DNase I stock, by adding it to the DNA 5 minutes before I added the cells, and found that it wasn't very effective - the residual transformation frequency was still quite high. This means that some cells might have taken up DNA while they were on the plate, but since this would only exacerbate the expression-time problem it shouldn't have been a big factor.
I only did this experiment because it would help us interpret the lack of effect of the HI0659 knockout on growth rates (in the experiment I described yesterday). If most cells weren't competent even in the hypercompetence mutants, then not seeing a growth defect in the culture doesn't meen that the competent cells didn't experience a growth defect. But this weird result means I need to do more experiments to figure out the reason for the discrepancy with previous results.
Usually when I do this kind of experiment I find that only some cells were competent (anywhere from about 10% to about 50%). But this time I got a very different result, and I don't know why.
I was testing three strains - wildtype cells (strain KW20) and two hypercompetent mutant derivatives, carrying the sxy1 and murE749 mutations. Both KW20 and sxy1 have been tested several times before; mutrE749 hasn't.
The procedure is simple. Transform cells with MAP7 chromosomal DNA and select for two antibiotic resistance mutations located far apart on the chromosome (so they won't ever be carried on the same DNA fragment). Select for each resistance separately and for the two together (double-transformants). Count the resulting colonies and calculate the transformation frequency for each resistance separately and for the double-transformants.
Calculate the fraction competent as the product of the two single-mutation transformation frequencies divided by the double-transformant frequency and by a fudge factor somewhere between 2 and 4.
The fudge factor incorporates two sub-factors accounting for different effects. The first effect is the chance that a single cell took up and recombined two DNA fragments, each containing one of the mutations, but that the incoming DNAs recombined with different strands so that, when the cell divided, one daughter cell got one mutation and the other got the other. This factor is complicated by the unknown effects of mismatch repair, but on average 2 is the appropriate value. The second effect is whether the cells did any cell divisions before they were placed on the agar to grow into colonies. If they did, then cells transformed by a single mutation will have given rise to one resistant colony and one sensitive colony. This factor should have a value of 2 if all the cells had time to divide before plating (if, for example, they needed time to express the antibiotic resistance), by less than 2 if only some did, and by 1 if the cells were plated immediately after transformation. In my experiment I didn't allow any expression time* so this second factor should be closer to 1 than 2. For simplicity I'll use a complete fudge factor of 3.
* In retrospect I should have allowed expression time, because one of the markers I was selecting for is spectinomycin resistance, which we think of as needing an hour's expression time. But I forgot and, rather surprisingly, still got tons of transformants.
Here are the calculations:

Might my assumptions be invalid? This analysis requires that cells be able to take up more than one fragment of DNA, and that taking up one fragment does not affect the probability of taking up another fragment. Because previous experiments have always given values less than 1, we've been assuming that this requirement is met. But this new result only makes sense if many of the cells could only take up one fragment of DNA.
Might my DNA or plates be faulty? The problem isn't due to using some new DNA prep with different properties; I used a fresh tube of the same MAP7 DNA stock we've been using for years. Could it be because I forgot to give the cells some expression time after DNA uptake? Might the lack of expression time reduced the numbers of double transformants (SpcR NovR) much more than it reduced the numbers of single SpcR transformants? But lack of expression time doesn't appear to have been a problem, since I got at least as many SpcR transformants as NovR transformants. The DNase I stock could be a problem. The transformation reactions are stopped after 15 minutes by adding DNase I to degrade the remaining DNA - a few days ago I tested the DNase I stock, by adding it to the DNA 5 minutes before I added the cells, and found that it wasn't very effective - the residual transformation frequency was still quite high. This means that some cells might have taken up DNA while they were on the plate, but since this would only exacerbate the expression-time problem it shouldn't have been a big factor.
I only did this experiment because it would help us interpret the lack of effect of the HI0659 knockout on growth rates (in the experiment I described yesterday). If most cells weren't competent even in the hypercompetence mutants, then not seeing a growth defect in the culture doesn't meen that the competent cells didn't experience a growth defect. But this weird result means I need to do more experiments to figure out the reason for the discrepancy with previous results.
the HI0660 'toxin' doesn't affect cell growth or survival
My last experiment showed that HI0660 encodes a 'toxin' of some sort, which prevents transformation when induced in the absence of the antitoxin encoded by HI0659. So yesterday I did detailed growth curves for cultures carrying the HI0659 knockout and either of our hypercompetence mutations.
The logic is that neither HI0660 or HI0659 will be expressed during growth in wildtype cultures, because the competence genes are not induced then. They're weakly induced at the end of growth, but strongly induced only when cells are starved or in the presence of the hypercompetence mutations. This means that, if HI0660 does kill cells or prevent growth, this effect will best be seen in the presence of the hypercompetence mutations.
The results show no evidence of significant growth or survival differences due to unopposed expression of HI0660. Each line in the two graphs below shows the mean for 7 replicate wells of the same inoculum. The upper graph used inocula that were of single small colonies diluted into 10 ml medium. The lower graph used inocula that were 1/1000 dilutions of overnight cultures into medium.
This is an interesting result, because it suggests that HI0660 acts directly on DNA uptake, not by killing cells or interfering with their growth. Because homologs of HI0660 are known to act by inactivating specific mRNAs, we may have to use RNA-seq to identify its mode of action.


The logic is that neither HI0660 or HI0659 will be expressed during growth in wildtype cultures, because the competence genes are not induced then. They're weakly induced at the end of growth, but strongly induced only when cells are starved or in the presence of the hypercompetence mutations. This means that, if HI0660 does kill cells or prevent growth, this effect will best be seen in the presence of the hypercompetence mutations.
The results show no evidence of significant growth or survival differences due to unopposed expression of HI0660. Each line in the two graphs below shows the mean for 7 replicate wells of the same inoculum. The upper graph used inocula that were of single small colonies diluted into 10 ml medium. The lower graph used inocula that were 1/1000 dilutions of overnight cultures into medium.
This is an interesting result, because it suggests that HI0660 acts directly on DNA uptake, not by killing cells or interfering with their growth. Because homologs of HI0660 are known to act by inactivating specific mRNAs, we may have to use RNA-seq to identify its mode of action.


Woo-hoo!! A hypothesis proved correct!
Last spring I came up with a far-fetched hypothesis to explain the phenotypes of two of our competence-gene knockouts,HI0569 (competence eliminated) and HI0660 (normal competence). I proposed that HI0660n encoded a 'toxin' that prevents competence or kills cells expressing it, and that HI0659 encodes an antitoxin that protects cells from the actions of HI0660. You can read all about it here.
Yesterday I finally was able to do the critical experiment, testing the competence phenotype of cells with both genes knocked out. It's normal!
And here's the data:


Yesterday I finally was able to do the critical experiment, testing the competence phenotype of cells with both genes knocked out. It's normal!
And here's the data:

The asterisk on the HI0659 column indicates that this is a 'less than' data point, since there were no transformant colonies on any of the plates.
This result confirms that HI0660 does something that COMPLETELY prevents transformation, and that HI0659's job is to prevent HI0660 from doing whatever it does.

hfq results
Yes indeed, hfq is needed for full competence development. The mutant grows as well as its wild-type parent (top graph) but develops about ten-fold lower competence (lower graph).

Next step: Make DNA from the mutant and use it to transform the hfq knockout into the hypercompetent mutants. Then I'll test the effect of the hfq knockout on their competence. If Hfq avcts directly on the sxy mRNA stem that regulates translation, I expect the mutants to be unaffected by loss of Hfq because their translation is not limited by the stem.
As a check that Hfq's effects aren't due to indirect effects on cAMP levels or CRP activity, I'll also test the effect of the knockout in wildtype and hypercompetent cells with added cAMP.
What about effects on the murE hypercompetence mutants? I'll test that, but I'm not sure how to interpret different results...
Subscribe to:
Posts (Atom)