














Last summer I started the blog post below.
Does bicyclomycin induce competence?
Yesterday the summer student pulled out the public data files for E. coli microarray experiments that had included measurements of sxy mRNA. We don't know how sxy expression is controlled in E.coli - nobody has found a way to induce expression of the chromosomal gene (we used an inducible plasmid clone to study its effects on other genes). So it's good to see that some treatments did induce it.
In the diagram below, each coloured vertical bar represents a single microarray comparison of sxy mRNA under two different conditions. Mousing over the bar brings up a box describing the comparison and results. Most of the bars are black or blackish; these are comparisons where sxy mRNA levels are the same. Yellow bars are ones where it is down (bright yellow is ≥ 8-fold down, and blue bars are ones where it is up (bright blue is ≥8-fold up) (the scale is 'log 2 expression ratio').
It's hard for me to tell which (if any) patterns are biologically significant. The one I'm excited about
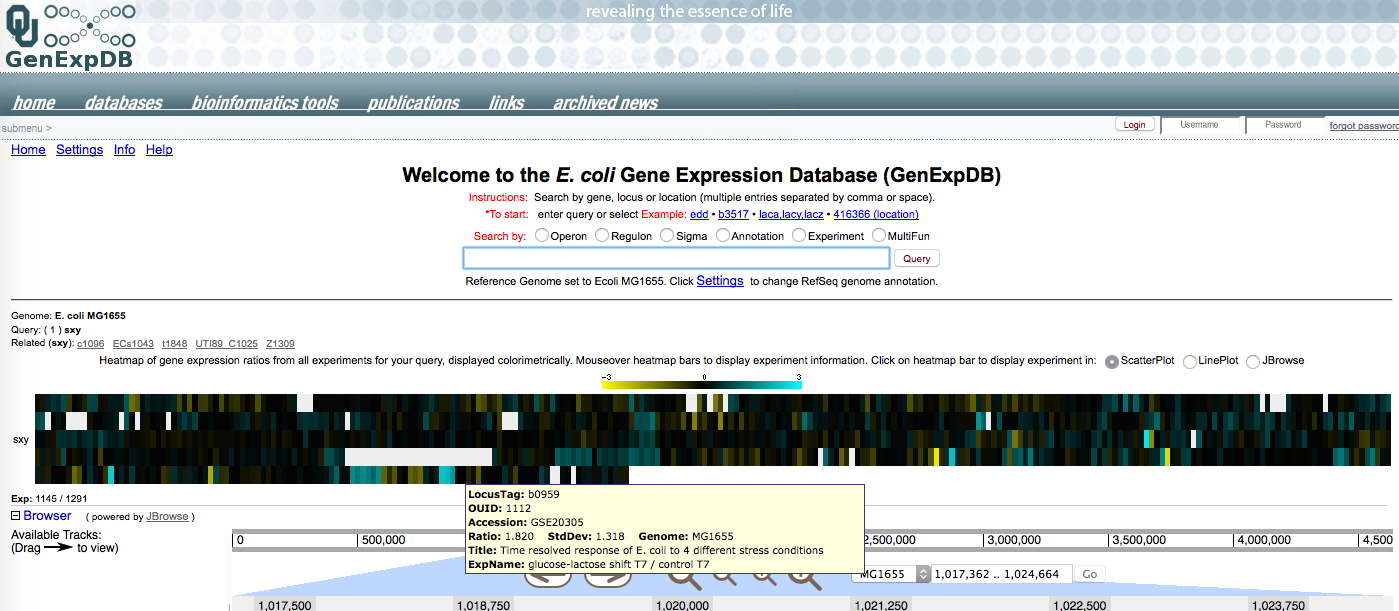
And that's the end of the draft post!
Subsequently I found a colleague who kindly gave me some bicyclomicin (it's an antibiotic), and roughed out a simple experiment. Now I'm planning to train up our new summer undergrad so she can do the experiment.
But I can't remember why I thought that bicyclomycin might induce competence!
Bicyclomycin is an antibiotic. I'd never heard of it until last summer, but it's of general interest because it's the only antibiotic that inhibits the Rho transcription termination protein. Given that competence development is limited by folding of the 5' end of sxy mRNA, it could be that Rho-mediated termination plays a role in determining whether sxy mRNA is translated.
Searching my blog posts for 'bicyclomycin' found the unpublished post above, which tantalizingly breaks off in mid-sentence just at the point where I was about to explain my interest. The figure is a screenshot from a microarray database, and I would expect that one of the bright-blue bars (sxy induction) would be from an array analysis involving bicyclomycin. But that doesn't seem to be the case. Of the five analyses with bright blue bars, one is UV irradiation, two are biofilms, one is heat shock, and one is a glucose-lactose shift. No mention of transcriptional termination. Searching the microarray database for 'bicyclomycin' brings up the expression of the bcl gene, whose mutations confer resistance, and a study of transcription termination in which sxy expression is unchanged!
This microarray study of transcription used bicyclomycin to inhibit termination. So I dug farther into it to see if there were any changes in expression of the competence-gene homologs that sxy induces. Some of them are tantalizingly up (the major T4P pilin and the comABCDE-homologs that specify the secretin pore and components of the T4P motor responsible for DNA uptake), but others are unchanged.
Subsequent searching also found an email I'd send to the summer student, with a link to this termination paper (Cardinale et al. 2008), asking 'Is this the one?". So I think this study is indeed what got me interested in bicyclomycin.
So let's see what the new summer student can find out!











