














I have a two-pronged plan to get a phage strain that gives good enough plaques for my GTA-as-vaccine experiments.
I obtained reasonable titers of two phages, 'Titan' and 'Saxon'. I'll invest a couple of weeks to see if I can get better and more reproducible plaques with either of these. The genome sequences of these phages are not closely related.
First, improve the plaquing conditions: The researcher who isolated the phages recommends using for the lawn cells that have been grown photosynthetically to a high density, He also suggested trying a lower top-agar concentration. I'll play around with these and other variables to see if I can get better plaques.
Second, use artificial selection to get a better strain of phage: I'll pick the few best-looking plaques of each of my two phages and plate the phage they contain in new lawns. From those new lawns I'll again pick the best-looking plaques, and plate their phage in new lawns. Etc. Maybe I'll introduce a bit of UV mutagenesis along the way.
The first step will be to make fresh lysates of these phages. The lawns I made before are too old, so I'll grow up some cells for lawns today and tomorrow I'll retiter the lysates. On Friday I can pick plaques from these lawns and make plate lysates. If there's a plate with near-confluent plaques I can use it directly to make a plate lysate. (10^7 or 10^8 pfu/ml, and I have maybe 5 µl so at best I can get plates with 5 x 10^4 or 5 x 10^5 plaques. The latter might be enough to get a good lysate. There are small volumes (50-100 µl?) of the original lysates in the lab upstairs, so maybe I'll sue these. Or Maybe I should save these until I've improved the plaquing conditions.
Field of Science
-
-
Change of address1 year ago in Variety of Life
-
Change of address1 year ago in Catalogue of Organisms
-
-
Earth Day: Pogo and our responsibility1 year ago in Doc Madhattan
thin lawns, feeble or absent phage
My phage titering gave disappointing results. Three of the five lysates gave no plaques at all, and the other two gave small indistinct plaques that couldn't be accurately counted or characterized.

I took some photos of the plaques I did see. The top photo is a section of one lawn, with several thousand tiny indistinct plaques. (The blurry markings are the label on the bottom of the plate.) The second photo is a closeup of an area on another lawn where I had spotted more-dilute phage, taken with my iPhone's Olloclip zoom lens. A few tiny plaques are visible, maybe 3, maybe 5.




I took some photos of the plaques I did see. The top photo is a section of one lawn, with several thousand tiny indistinct plaques. (The blurry markings are the label on the bottom of the plate.) The second photo is a closeup of an area on another lawn where I had spotted more-dilute phage, taken with my iPhone's Olloclip zoom lens. A few tiny plaques are visible, maybe 3, maybe 5.


For comparison, here's what nice plaques look like. These are plaques of the E. coli phage lambda (source)

I won't be able to use these R. capsulatus phage for my GTA-vaccine experiments unless I can get better plaques. I'll need to know whether the phage makes turbid or clear plaques, and I'll need to be able to count it accurately.
I can try using another strain as the host. These lawns were made with a culture of strain YW1, the strain that these phage were originally isolated on. I have several other strains, though I don't know if they are closely related.
I can also try changing the plating conditions. I followed the protocol that I obtained from people who have worked with these phage, but perhaps I could grow the cells to a different density, or incubate the plates at a different temperature. I'll ask the experts for advice.
Titering my lysates
Planning today's work:
Titering the phage lysates should be a no-brainer, but it's been a long time since I worked with phage so I'd better think things through before I do it.
I have 15 µl of each of 5 phage stocks ('lysates'). The original titers (plaque-forming units/ml, pfu/ml) are written on the tubes - they range from 6x10^5 pfu/ml to 2x10^11 pfu/ml. But the lysates are probably quite old (maybe 2 years, maybe more), so their titers may have dropped a lot.
I think I'll do one poured-lawn plate of each, using an amount of lysate that should be about 1000 pfu according to the original titer. And I'll do a spotted-lawn plate for each phage, using undiluted lysate and a range of dilutions.

I'll dilute the lysates in the same YPS medium I've grown the cells in. It has calcium and magnesium added so should be fine for typical phage.
Titering the phage lysates should be a no-brainer, but it's been a long time since I worked with phage so I'd better think things through before I do it.
I have 15 µl of each of 5 phage stocks ('lysates'). The original titers (plaque-forming units/ml, pfu/ml) are written on the tubes - they range from 6x10^5 pfu/ml to 2x10^11 pfu/ml. But the lysates are probably quite old (maybe 2 years, maybe more), so their titers may have dropped a lot.
I think I'll do one poured-lawn plate of each, using an amount of lysate that should be about 1000 pfu according to the original titer. And I'll do a spotted-lawn plate for each phage, using undiluted lysate and a range of dilutions.

I'll dilute the lysates in the same YPS medium I've grown the cells in. It has calcium and magnesium added so should be fine for typical phage.
About bacterial lawns and phage plaques
This was going to be a post where I do the planning to titer my new lysates today, but it turned into an explanation of how microbiologists use plaques in lawns of bacteria to study phages.
Wait, what's a 'lawn' and what's a 'plaque'? A lawn is a thin layer of confluent bacterial growth, usually created by mixing a relatively large number of cells (≥10^6) with liquid agar solution ('top agar' or 'soft agar' and pouring the mixture onto the surface of a nutrient-containing agar plate. The top agar is usually at 0.5-0.75%, about half the concentration used for a normal solid plate. The cells can't move around in the agar, and they grow to high density using the nutrients that diffuse upward from the bottom layer.


Here is a detailed drawing of what's happening as a plaque forms:
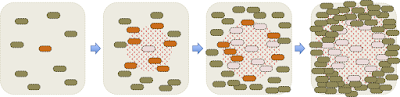
Initially there's just one infected cell, and sparse uninfected cells in its neighbourhood. When this cell lyses, the phages it releases can readily diffuse through the agar and infect nearby cells. While this is happening, the uninfected cells are growing and dividing. When the newly infected cells lyse, the phage they release add to the local population and infect more cells. The phage continue to diffuse away, but soon the neighbouring cells become so dense that they stop growing and the phage can no longer replicate in them. The cells are too big to diffuse through the agar like the phage, so lysis leaves a circular cell-free space called a plaque. Typical plaques are 1-2 mm across so easy to see with the naked eye.
By counting the number of plaques that form in a lawn of bacteria, we know how many infectious phages were present in the mixture we poured on the plate. This is the standard way to measure the number of phages (well, the number of 'plaque forming units', PFUs) in a preparation of phage (a 'lysate').
Regular poured-lawn method: Cells and diluted phage are incubated together in a small volume of liquid (broth or phage-dilution solution) for long enough that most of the phage have attached to the cells. Then hot liquid top agar is added to the tube and the contents are quickly mixed and poured onto an agar plate of whatever medium best supports lawn growth and plaque formation. (Quickly so the mixture cools before the cells are damaged.) The top agar quickly sets, and the plate is incubated overnight at an appropriate temperature for bacterial growth and phage plaque formation.
Spot-titer method: Cells are quickly mixed with hot top agar (no phage) and the mixture is poured onto agar plates and left to set. Sometimes the plates can be prepared days in advance, if the cells are happy sitting in the fridge. 10 µl dilutions of phage are then spotted onto the surface and the plates are incubated overnight as before. If you're gentle you can even streak a drop of lysate across the lawn as you would streak cells on a normal plate, allowing you to grow well-isolated plaques without the nuisance of diluting your lysate.
Other ways we can use lawns and plaques:
Isolating phage from a single plaque: Often you want to start an experiment with a genetically pure phage lysate that you grew up from a single plaque. If plaques are well-separated (remember that the phage continue to diffuse out after the plaque forms and the lawn stops growing) you can use a Pasteur pipette to punch out the plaque away from the surrounding agar. If this plaque is put into a small volume of phage-dilution solution, the many thousands of phages it contains will diffuse out over a few hours (or less) and the phage-containing liquid can be used in your experiment, or to prepare a new lysate whose phage all derive from the one that originated the plaque.
Plate lysates: Lysates can be prepared in broth, by adding phage to growing cells, watching for the time when the culture clears because most of the cells have lysed, and pelleting out the cell debris. This is a bit fussy to do, since clearing depends on having the right proportions of phage and cells. A simpler method is to prepare a 'plate lysate', as follows. Mix the liquid from a picked plaque or a small amount of a lysate with cells and top agar, and pour a lawn. You want enough phage that the resulting plaques will be 'confluent' - will overlap just enough that very little intact lawn remains. Once the plaques have formed, overlay the top agar with 5 ml of phage-dilution solution and leave for a few hours or overnight. Half of the phage will diffuse into the liquid, and in the morning you just have to collect the liquid and add a few drops of chloroform to kill any cells. These lysates usually have very high titers, because the cells in a lawn can grow to much higher density than those in a liquid culture.
Phage-resistant colonies: Sometimes, the area around an initially infected cell includes a cell that is genetically resistant to the phage due to a new mutation that blocks phage attachment or reproduction. Such as cell (green in the diagram below) will be able to grow within the area of spreading phage, and its descendants will form a visible colony within the plaque.

Turbid plaques: One other phenomenon deserves mention, and that's the 'turbid' plaques formed when a 'temperate' phage infects a lawn. Temperate phages are those that have a mechanism to enter a dormant state in host cells, where the phage genome is passively replicated by the cellular machinery, usually because it is integrated into the cell's chromosome. Cells with such dormant phages ('lysogens', orange in the diagram below) are resistant to infection by external phages. When a temperate phage forms a plaque, most infected cells lyse and produce infectious phage, but some form lysogens that grow and divide within the plaque. Usually many such cells form causing the center of the plaque to appear cloudy ('turbid') rather than having visible colonies.

Wait, what's a 'lawn' and what's a 'plaque'? A lawn is a thin layer of confluent bacterial growth, usually created by mixing a relatively large number of cells (≥10^6) with liquid agar solution ('top agar' or 'soft agar' and pouring the mixture onto the surface of a nutrient-containing agar plate. The top agar is usually at 0.5-0.75%, about half the concentration used for a normal solid plate. The cells can't move around in the agar, and they grow to high density using the nutrients that diffuse upward from the bottom layer.

If a few of the initial cells were infected with a phage, the phage they release when they die will infect neighbouring cells and kill them, creating a cell-free zone called a 'plaque'.

Here is a detailed drawing of what's happening as a plaque forms:

By counting the number of plaques that form in a lawn of bacteria, we know how many infectious phages were present in the mixture we poured on the plate. This is the standard way to measure the number of phages (well, the number of 'plaque forming units', PFUs) in a preparation of phage (a 'lysate').
Regular poured-lawn method: Cells and diluted phage are incubated together in a small volume of liquid (broth or phage-dilution solution) for long enough that most of the phage have attached to the cells. Then hot liquid top agar is added to the tube and the contents are quickly mixed and poured onto an agar plate of whatever medium best supports lawn growth and plaque formation. (Quickly so the mixture cools before the cells are damaged.) The top agar quickly sets, and the plate is incubated overnight at an appropriate temperature for bacterial growth and phage plaque formation.
Spot-titer method: Cells are quickly mixed with hot top agar (no phage) and the mixture is poured onto agar plates and left to set. Sometimes the plates can be prepared days in advance, if the cells are happy sitting in the fridge. 10 µl dilutions of phage are then spotted onto the surface and the plates are incubated overnight as before. If you're gentle you can even streak a drop of lysate across the lawn as you would streak cells on a normal plate, allowing you to grow well-isolated plaques without the nuisance of diluting your lysate.
Other ways we can use lawns and plaques:
Isolating phage from a single plaque: Often you want to start an experiment with a genetically pure phage lysate that you grew up from a single plaque. If plaques are well-separated (remember that the phage continue to diffuse out after the plaque forms and the lawn stops growing) you can use a Pasteur pipette to punch out the plaque away from the surrounding agar. If this plaque is put into a small volume of phage-dilution solution, the many thousands of phages it contains will diffuse out over a few hours (or less) and the phage-containing liquid can be used in your experiment, or to prepare a new lysate whose phage all derive from the one that originated the plaque.
Plate lysates: Lysates can be prepared in broth, by adding phage to growing cells, watching for the time when the culture clears because most of the cells have lysed, and pelleting out the cell debris. This is a bit fussy to do, since clearing depends on having the right proportions of phage and cells. A simpler method is to prepare a 'plate lysate', as follows. Mix the liquid from a picked plaque or a small amount of a lysate with cells and top agar, and pour a lawn. You want enough phage that the resulting plaques will be 'confluent' - will overlap just enough that very little intact lawn remains. Once the plaques have formed, overlay the top agar with 5 ml of phage-dilution solution and leave for a few hours or overnight. Half of the phage will diffuse into the liquid, and in the morning you just have to collect the liquid and add a few drops of chloroform to kill any cells. These lysates usually have very high titers, because the cells in a lawn can grow to much higher density than those in a liquid culture.
Phage-resistant colonies: Sometimes, the area around an initially infected cell includes a cell that is genetically resistant to the phage due to a new mutation that blocks phage attachment or reproduction. Such as cell (green in the diagram below) will be able to grow within the area of spreading phage, and its descendants will form a visible colony within the plaque.

Turbid plaques: One other phenomenon deserves mention, and that's the 'turbid' plaques formed when a 'temperate' phage infects a lawn. Temperate phages are those that have a mechanism to enter a dormant state in host cells, where the phage genome is passively replicated by the cellular machinery, usually because it is integrated into the cell's chromosome. Cells with such dormant phages ('lysogens', orange in the diagram below) are resistant to infection by external phages. When a temperate phage forms a plaque, most infected cells lyse and produce infectious phage, but some form lysogens that grow and divide within the plaque. Usually many such cells form causing the center of the plaque to appear cloudy ('turbid') rather than having visible colonies.

Questions about CRISPR-mediated phage immunity
Thursday's post described the hypothesis that bacteria might use gene transfer agent particles to inoculate other cells in the population with fragments of phage DNA, and outlined an experiment to test this. Now I'm realizing that I need to know a lot more about the kind of immunity I should expect to see if this GTA-as-vaccine hypothesis is correct.

Simplistic outline of the experiment:
Things I should find out before I do the experiment:
1. How efficiently do introduced DNA fragments give rise to CRISPR spacers? If this efficiency is too low relative to the background rate of phage resistance, I won't be able to detect an effect. This paper (Hynes et al. 2014, thanks to @AprilPawluk for pointing me to it) might let me estimate the efficiency. They exposed cells to a mixture of infectious and damaged phage (damaged by a restriction enzyme in the cell or by prior UV irradiation) at a multiplicity of infection (moi) of 0.1-0.2, and then examined the resulting confluently lysed lawns for phage-resistant colonies. Unfortunately they only report relative changes in frequency of resistant cells (maxima 16-fold and 6 fold for restriction and irradiation respectively), but in their Methods they mention that the highest frequencies of resistance they observed were about 10^-6. I don't know if this is for naive cells or pre-exposed cells, but even if it's for naive cells, the max frequency of CRISPR resistance I might expect is only about 10^-5. This would not pose a detection problem, but it would limit the population-level benefits of the proposed vaccine system.
2. What fraction of the survivors of a phage infection are genetically resistant, and what fraction of phage resistance arises by non-CRISPR mechanisms? If most survivors are just lucky, then it might be a lot of work to identify the genetically resistant ones. In the Hynes et al. experiments, all of the colonies were genetically resistant, and all had new CRISPR spacers. However this might be quite different for different phages. If most resistant cells have altered phage receptors rather than phage-specific CRISPR spacers, the effect of GTA-mediated CRISR resistance will be hard to detect.
3. How quickly does CRISPR-mediated phage resistance arise after exposure to phage DNA? I don't know. Cells in the Hynes experiment might have had one or two lytic-cycle durations between being infected by the damaged phage and being infected by an infectious phage.
4. What fraction of phage infections are abortive and thus could lead to CRISPR immunity to subsequent infection? Inspired by the Hynes experiment, I can increase abortive infections by UV-irradiating the phage lysate. (I know how to do this well from previous work.)
5. How efficiently do phage spacers prevent phage infection? April Pawluk (via Twitter) says they reduce infection by several orders of magnitude. In the Hynes work acquisition of a phage-derived CRISPR spacer enabled cells to form a colony in a sea of phage.
OK. I have lysates of five sequenced R. capsulatus phages (from Dave Bollivar via Tom Beatty), and I have the R. capsulatus strain these phages were isolated on, as well as GTA-producing and recipient strains. Time to get to work!

Simplistic outline of the experiment:
- Infect GTA-producer strain of R. capsulatus with phage under conditions where the infection is inefficient and few cells lyse.
- Remove cells and debris from the culture, to get a supernatant that will contain GTA particles and (unavoidably) some phage particles.
- Expose a new culture to the supernatant so cells obtain DNA from the GTA particles, again under conditions where successful phage infection will be minimized.
- Wash the surviving cells to remove phage (as much as possible). Allow time for CRISPR formation if needed.
- Expose the cells to a titer of phage suitable for selecting resistant cells. As a control, also expose cells not treated with GTA.
- Plate to isolate colonies from surviving cells.
- Test the survivors for phage resistance.
- Compare the frequency of resistance in treated and control cultures.
- Test resistant colonies for CRISPR changes.
Things I should find out before I do the experiment:
1. How efficiently do introduced DNA fragments give rise to CRISPR spacers? If this efficiency is too low relative to the background rate of phage resistance, I won't be able to detect an effect. This paper (Hynes et al. 2014, thanks to @AprilPawluk for pointing me to it) might let me estimate the efficiency. They exposed cells to a mixture of infectious and damaged phage (damaged by a restriction enzyme in the cell or by prior UV irradiation) at a multiplicity of infection (moi) of 0.1-0.2, and then examined the resulting confluently lysed lawns for phage-resistant colonies. Unfortunately they only report relative changes in frequency of resistant cells (maxima 16-fold and 6 fold for restriction and irradiation respectively), but in their Methods they mention that the highest frequencies of resistance they observed were about 10^-6. I don't know if this is for naive cells or pre-exposed cells, but even if it's for naive cells, the max frequency of CRISPR resistance I might expect is only about 10^-5. This would not pose a detection problem, but it would limit the population-level benefits of the proposed vaccine system.
2. What fraction of the survivors of a phage infection are genetically resistant, and what fraction of phage resistance arises by non-CRISPR mechanisms? If most survivors are just lucky, then it might be a lot of work to identify the genetically resistant ones. In the Hynes et al. experiments, all of the colonies were genetically resistant, and all had new CRISPR spacers. However this might be quite different for different phages. If most resistant cells have altered phage receptors rather than phage-specific CRISPR spacers, the effect of GTA-mediated CRISR resistance will be hard to detect.
3. How quickly does CRISPR-mediated phage resistance arise after exposure to phage DNA? I don't know. Cells in the Hynes experiment might have had one or two lytic-cycle durations between being infected by the damaged phage and being infected by an infectious phage.
4. What fraction of phage infections are abortive and thus could lead to CRISPR immunity to subsequent infection? Inspired by the Hynes experiment, I can increase abortive infections by UV-irradiating the phage lysate. (I know how to do this well from previous work.)
5. How efficiently do phage spacers prevent phage infection? April Pawluk (via Twitter) says they reduce infection by several orders of magnitude. In the Hynes work acquisition of a phage-derived CRISPR spacer enabled cells to form a colony in a sea of phage.
OK. I have lysates of five sequenced R. capsulatus phages (from Dave Bollivar via Tom Beatty), and I have the R. capsulatus strain these phages were isolated on, as well as GTA-producing and recipient strains. Time to get to work!
Why GTA genes can't be maintained by 'selfish' transmission
Below is the line of reasoning showing that the genes responsible for producing GTA particles cannot maintain themselves or spread into new populations by GTA-mediated transfer of themselves into new cells. I initially worked this out with a rigorous set of mathematical equations, but then realized that the problem was so glaringly obvious that math isn't needed.
The main GTA gene cluster is too big to fit inside a single GTA particle, so GTA particles can't transmit DNA that converts a GTA- cell into a GTA+ cell. Some genes outside the main cluster are also required for GTA production.



The main GTA gene cluster is too big to fit inside a single GTA particle, so GTA particles can't transmit DNA that converts a GTA- cell into a GTA+ cell. Some genes outside the main cluster are also required for GTA production.

But GTA particles can (and do) contain one or more individual GTA genes. If a fragment containing a particular GTA gene is injected into a formerly-GTA+ cell that is now GTA- because it has a mutated version of this gene, the resulting recombination can restore the cell's original GTA+ genotype.

But these transfer events would not allow GTA+ cells to invade a GTA- population, or to maintain themselves in the face of loss of GTA function by mutation. That's true for all known GTA systems, even in the simplest (imaginary) case where production of GTA particles requires only a single gene that could easily fit into a GTA particle, as illustrated below.

Why? Three factors together require that production of GTA particles reduces the total number of GTA+ cells in the population:
Problem 1: GTA particles can only be released to the environment if the GTA+ producer cell lyses. So each production event removes one GTA+ cell from the population.
Problem 2: The GTA genes in the producer cell are not over-replicated as a phage genome would be, so each production event can produce at most one G+ particle (containing the GTA gene or cluster).
If all steps occurred with 100% efficiency, problems 1 and 2 would allow, at best, replacement of the lost GTA+ cell with a new one created by GTA-mediated recombination. But this would not maintain the numbers of GTA+ cells in the face of occasional loss of GTA genes by mutation or deletion. Nor would it allow GTA+ cells to invade a GTA- population.
Problem 3: Production of GTA particle production, transmission of their DNA to recipient cells, and recombination with the recipient genome are all likely to be at least moderately inefficient. Here's a partial list of expected inefficiencies:
- Burst size: Actual burst sizes are unknown, but packaging all the DNA in a R.capsulatus. genome would need 841 particles, which is much larger than typical burst sizes for DNA phages. Capsid proteins may be limiting, since they would be produced from single-copy GTA genes rather than replicated phage genomes.
- Dispersion: The GTA particles will disperse in the environment, and many will probably not find cells to attach to.
- Stability: Lab preps of GTA particles are unstable in non-optimal storage conditions, so many particles will likely fall apart.
- Recombination efficiency: Only one DNA strand enters the cytoplasm, and some DNA degradation is likely. The highest observed transduction frequency is only ~4^-4, (theor. max: 1.2^-3) so recombination efficiency is probably only ~0.3. Recombining in a novel gene will be less efficient than simple strand replacement
- Self-conversion: Some G+ particles may attach to cells that are already GTA+.
Might GTA be a vaccination system for infecting phages?
My work at Dartmouth (to be described in upcoming posts) showed conclusively that genes encoding Gene Transfer Agents (such as the GTA system of Rhodobacter capsulatus) cannot be maintained by 'selfish' transfer of either whole GTA gene clusters or single GTA genes into GA- recipients. Neither can the GTA genes be maintained by general recombination benefits that can arise when fragments of chromosomal DNA are transferred into new cells. So, although 'gene transfer agent' does accurately describe one activity of these genes, it cannot be the activity for which they are selected.

The main obstacle to the maintenance of GTA genes, which applies to all the benefits is that any GTA+ cell that actively produces GTA particles cells must die, since cell lysis is needed to release their particles into the environment. Another obstacle, applying to selfish transfer, is that GTA genes are not over-replicated during GTA production (and are not preferentially packaged), so each cell death can produce only one GTA+ particle.

I presented these results at the Analytical Genetics conference last week, and asked the other participants if they could think of alternative benefits of producing GTA particles. Sanna Koskiniemi from Uppsala University made the very interesting suggestion that GTA particles could serve as a syringe, packaging DNA fragments from a phage that's infecting the producer cell and transferring these fragments into other as-yet-uninfected cells, where they could trigger development of CRISPR immunity.
I love this idea and want to test it. It doesn't overcome the cell-death obstacle, but it does overcome the selfish-transfer obstacle since a single producer cell could produce many particles of phage DNA from a single phage genome, and more if the phage genome is replicated.

One way to see if this could provide sufficient benefits to maintain the GTA genes is by simulation modeling like that I used to examine the recombination benefits. This could clairfy the important factors that would need to be examined.
Here I want to start considering experimental tests of this hypothesis.
The ideal test would be to infect the GTA-producing strain with a phage, preferably under low-growth conditions where phage infections are often abortive. (Luckily R. capsulatus produces most of its GTA under such conditions.) Then some recipient cultures would be exposed to the GTA-containing culture medium (and some not, as controls), and then all exposed to a lysate of the phage.
"But wait!", you say. "Won't the GTA-containing culture medium also contain some phage?" Yes, probably. I don't think there's any way to inactivate the phage particles without also inactivating the GTA particles, or vice versa. We might be able to come up with either perfectly-abortive infection conditions (where infected cells don't produce any phage), or a cellular mutation that prevents phage production. If not, we might have to combine the GTA-exposure and phage-infection steps.
"And won't any phage lysate also contain some GTA particles?" Yes, probably. But we could use a GTA- mutant as the host for lysate production. Not the mutant that can't lyse, but the one with the main GTA gene cluster completely deleted.
What resources are available for this project? First I checked with my GTA colleagues, who confirm that R. capsulatus does have a CRISPR-Cas9 system. Then I asked if there were any well-characterized phage systems able to infect R. capsulatus. Until quite recently the answer would have been 'No', but a recent paper reported the isolation and sequences of 4 R. capsulatus phages. A Mu-like phage of R. capsulatus has also been characterized, but it did not form plaques on SB1003.
The report about the 4 new phages used a different host strain (YW1-derived, not SB1003), so the first thing I'll need to do is check whether they form plaques on SB1003. Then I'll need to play around with infection and plating conditions... My idea of fun!


I presented these results at the Analytical Genetics conference last week, and asked the other participants if they could think of alternative benefits of producing GTA particles. Sanna Koskiniemi from Uppsala University made the very interesting suggestion that GTA particles could serve as a syringe, packaging DNA fragments from a phage that's infecting the producer cell and transferring these fragments into other as-yet-uninfected cells, where they could trigger development of CRISPR immunity.
I love this idea and want to test it. It doesn't overcome the cell-death obstacle, but it does overcome the selfish-transfer obstacle since a single producer cell could produce many particles of phage DNA from a single phage genome, and more if the phage genome is replicated.

One way to see if this could provide sufficient benefits to maintain the GTA genes is by simulation modeling like that I used to examine the recombination benefits. This could clairfy the important factors that would need to be examined.
Here I want to start considering experimental tests of this hypothesis.
The ideal test would be to infect the GTA-producing strain with a phage, preferably under low-growth conditions where phage infections are often abortive. (Luckily R. capsulatus produces most of its GTA under such conditions.) Then some recipient cultures would be exposed to the GTA-containing culture medium (and some not, as controls), and then all exposed to a lysate of the phage.
"But wait!", you say. "Won't the GTA-containing culture medium also contain some phage?" Yes, probably. I don't think there's any way to inactivate the phage particles without also inactivating the GTA particles, or vice versa. We might be able to come up with either perfectly-abortive infection conditions (where infected cells don't produce any phage), or a cellular mutation that prevents phage production. If not, we might have to combine the GTA-exposure and phage-infection steps.
"And won't any phage lysate also contain some GTA particles?" Yes, probably. But we could use a GTA- mutant as the host for lysate production. Not the mutant that can't lyse, but the one with the main GTA gene cluster completely deleted.
What resources are available for this project? First I checked with my GTA colleagues, who confirm that R. capsulatus does have a CRISPR-Cas9 system. Then I asked if there were any well-characterized phage systems able to infect R. capsulatus. Until quite recently the answer would have been 'No', but a recent paper reported the isolation and sequences of 4 R. capsulatus phages. A Mu-like phage of R. capsulatus has also been characterized, but it did not form plaques on SB1003.
The report about the 4 new phages used a different host strain (YW1-derived, not SB1003), so the first thing I'll need to do is check whether they form plaques on SB1003. Then I'll need to play around with infection and plating conditions... My idea of fun!
Subscribe to:
Posts (Atom)