














The problem and the strategy: We want to know how efficiently different segments of NP or GG DNA are taken up by competent Rd cells. All of our 12 'uptake' samples are contaminated, consisting of mostly reads of NP or GG DNA taken up by Rd cells plus varying amounts of contaminating Rd chromosomal DNA. We want to calculate the 'uptake ratio' for each genome position as the ratio of sequence coverage in the uptake sample to coverage in the 'input' sample - thus correcting for the varying efficiency of sequencing at different genome positions. We originally tried to identify and remove the Rd-derived reads from the uptake samples before calculating the uptake ratio, but this introduced new problems. Our new strategy is to instead deliberately add 'fake-contaminating' Rd reads to our input samples, at levels matching the real contamination in each uptake sample.
The test: To test whether the new strategy works, the PhD student first created a set of four fake-uptake samples by adding 10% of Rd reads (set 1) to each of the four input samples (NP-long, NP-short, GG-long, GG-short). He then created the corresponding fake-contaminated input samples by adding different Rd reads (set 2) to get the same 10% contamination level. He then calculated and plotted the ratio of fake-uptake to fake-input for each genome position. If the contamination correction were perfect this would give a value of 1.0 at every position, but we knew it would be imperfect because the contamination reads (set 1) and the correction reads (set 2) were not identical.
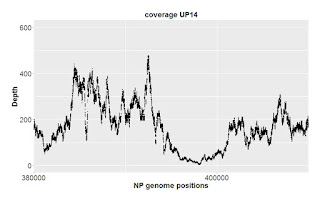
Here are the results for the NP-long analysis (the NP-short was similar):


Here are results for GG-long:





We should go back and test the same GG-long sample using different sets of Rd reads (set 3 rather than set 1, or set 4 rather than set 2).
Sources of variation: The above analysis gives us a better understanding of the sources of variation in this uptake analysis. First there's the variation across the genome in sequencing efficiency. This is (we think) due to properties of the sequencing technology, and should be constant across different samples from the same genome (e.g. input and uptake samples). We don't have any way to reduce this variation, but we control for it by calculating the uptake ratios rather than just position-specific differences in coverage in the uptake samples. Second, there's the variation in how much Rd contamination is present. This arises due to variation in the experiments that purified the DNA; we can't change it at this stage, but we control for the differences between samples by introducing different amounts of compensating fake-contamination into the input sample control for each uptake sample. Third, there's the chance variation in the distribution of contaminating Rd reads across the genome. This will be different for each sample, and we can't change it or control for it. Finally there's the random variation in the distribution of fake-contaminating Rd reads added to each input sample. The next section describes how we can eliminate most of this.
Replicate corrections will reduce variation: The above analysis also showed us a way to improve the correction in our 12 genuinely contaminated samples. Instead of doing each correction once, we can do the correction several times, generating independent fake-contamination input samples using independent sets of Rd reads. Then we can average the resulting uptake ratios. In fact we can do this a simpler way, by just averaging the coverage levels of the independent fake-contamination input samples at each position, and then calculating the uptake ratios using these averages.
The number of Rd reads needed for each correction will depend on the coverage level and true contamination level of each uptake sample, but we should have enough Rd reads to create at least
No comments:
Post a Comment
Markup Key:
- <b>bold</b> = bold
- <i>italic</i> = italic
- <a href="http://www.fieldofscience.com/">FoS</a> = FoS