We want to understand why competent
Haemophilus influenzae cells take up some parts of
H. influenzae chromosomes more efficiently than others.
To this end, before Christmas the grad student reisolated preparations of DNA fragments of chromosomal DNA from strain 26-028NP (hence 'NP') that had been taken up by competent cells of the standard lab strain Rd. He sent these DNA samples to the former post-doc for sequencing (with the original 'input DNAs as controls). The post-doc has now sent us the sequencing data, and the grad student is going to analyze this, with two main goals:
- Determine how a DNA fragment's probability of uptake is affected by the presence of sequences matching the uptake signal sequence ('USS') motif.
- Identify other sequence factors that influence uptake.
The grad student has written up an
overview of his plan for accomplishing these goals, and that has stimulated me to also think about how it could be done.
He (or the former post-doc?) has already done the first step, scoring the degree of preferential uptake for every position in the genome. I think this was done by comparing each genome position's coverage in the recovered-DNA dataset to its coverage in the control 'input' dataset. This gives a score they call the 'uptake ratio'.
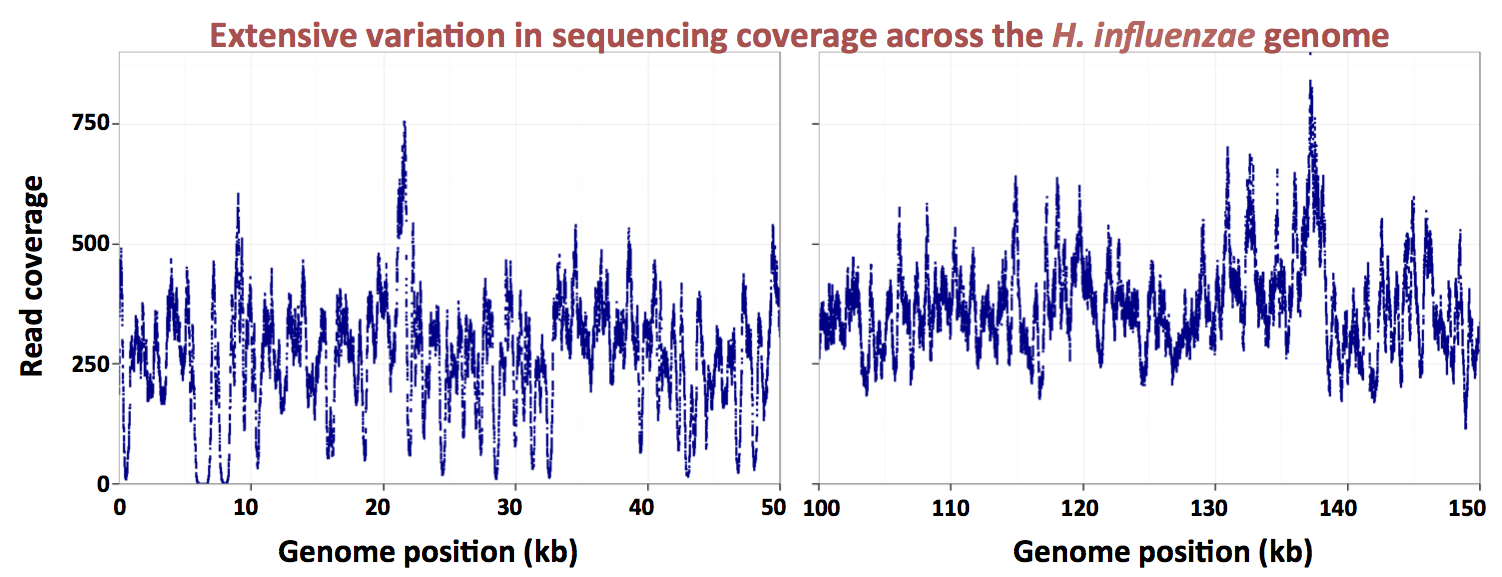
Here's a graph made by the grad student, showing the uptake ratios for two different preps of chromosomal DNA, over a 13 kb segment of the 1830 kb
H. influenzae Rd chromosome. The dark blue points are for a DNA prep whose average fragment size was about 6 kb, and the red points for a DNAS prep whose average fragment size was about 250 bp. Because the actual distributions of fragment sizes in these preps have not yet been carefully measured, I'll refer to them as the large-fragment and small-fragment DNA preps respectively.
The first thing you notice is that the uptake ratios for the large-fragment prep are much less variable than those for the small-fragment prep. We are very gratified to see this, because it's what we expected from the known contribution of the uptake motif. Sequences with strong matches to this motif occur all around the chromosome, with an average spacing of about 1 kb. Thus most fragments in the large-fragment prep will have contained at least one USS, but many fragments in the small-fragment prep will not have contained any USS.
The large-fragment prep does show two strong spikes of high uptake (at about 8000 and 18500 bp). These are certainly very interesting, especially since they don't correspond to high uptake in the short-fragment prep. But for now I'm just going to consider how we might analyze the short-fragment prep, since this provides much better resolution of what we think are the effects of individual USSs.
Here's a strategy I came up with:
Step 1: Score each position of the NP genome for its match to either orientation of the 'genomic' USS motif. This motif was identified by Gibbs Motif Sampler analysis of the RD genome (see
this paper). Each position will have a '+' score and a '-' score; we need to make sure the positions are aligned at the most important of the USS motif. Because the score depends on correct alignment, the result will be punctate, with about one high-scoring position and about 999 low-scoring positions in each kb. Here's a figure of what the analysis might look like for the 13 kb segment shown above.
Step 2: Using a reasonable score cutoff, create a list of positions that qualify as 'USS' for the initial analysis of the uptake data. In the case above we'd include all positions scoring higher than 15.
Step 3: For each position in the genome, calculate its distance from the nearest 'USS' on the above list. For now don't distinguish between 'USS' in + or - orientations. (I'm keeping 'USS' in quotes to remind us that we used only one of many possible criteria to define our list.)
Step 4: For each genome position plot its uptake ratio as a function of its distance from a 'USS'. Because most of the red peaks in the grad student's graph have uptake-ratio scores of about 4 and bases about 1 kb wide, I expect the graph to look something like this:
There are a lot more points on this graph than on the previous one because there's a point for every position in the 1.8 Mb genome. Most of the points fall on a rough band that drops from uptake ratios of 4 (peaks, for a very close 'USS') to uptake ratios that are about 0.1 (troughs, for positions that are more than 500 bp from a 'USS').
If we see a broad band with lots of scatter, this will mean that our distance-to-the-nearest-'USS' score doesn't capture other aspects of the USS that influence uptake. These factors might include:
- whether the USS's orientation on the chromosome affects uptake (USS motifs are asymmetric)
- how well the USS's sequence matches the several different ways we can score sequences as possible USS (genome-based, uptake-based, and with or without internal interaction effects between positions)
- how much the presence and relative locations of additional USSs adds to uptake
We will come back to the above analysis and develop more nuanced measures of the affects of nearby USS, judging success by how much each nuance reduces the scatter of the points.
For now I'm more interested in identifying any non-USS sequence factors that influence uptake. These factors should appear in the above graph as outliers, positions whose uptake ratio is not correlated with their USS-distance score. Our previous analysis suggests that these outliers should be common. If they are common, they might be clustered as shown above, but they're probably more likely to be scattered all over the place and perhaps not easily distinguishable from the overall background scatter.
The best way to see if these positions are not noise is to see if their scores correlate with genomic positions. Below is one way I've thought of to do this.
Step 5. Use the uptake vs USS-distance graph to develop an equation that best predicts uptake ratio (U) as a function of distance to nearest 'USS' (D, in bp). For the above example, a very crude equation might be
U = 0.1 or (4 - D/100), whichever is greater.
Step 6: For each position in the genome, use this equation and the 'USS' list to predict an uptake ratio, and then calculate the difference between its predicted and observed uptake ratios.
Step 7: Now plot this 'anomaly' as a function of genome position. If we're lucky it will look something like this:
If some of the apparent scatter is due to positions where non-USS sequences influence uptake, these will show up as peaks and troughs above and below the main bands, and we can go on to analyze these sequences bioinformatically for shared features and experimentally for direct effects on uptake. If the scatter really is due to noise, then it will be scattered over the genome and not fall into discrete peaks and troughs.