(In this post I'm only addressing the specific points made in this Q&A. Magenta numbers identify points I have concerns about. As always, I'm not asking readers to take my word for anything; if you have doubts you should look up the information and check my calculations for yourself.)
Question:
Some people have questioned whether the DNA was sufficiently cleaned by your technique using gel electrophoresis, to separate it from other molecules. Do you feel this is a valid concern?
Answer:
Our DNA extraction and purification protocol begins with washed cells, pelleted from media. These are then subjected to a standard DNA extraction protocol (1), which included multiple phenol chloroform steps to remove impurities, including any unincorporated arsenate (As) (2). After this, the DNA was electrophoresed, further separating the DNA from impurities (3, 4). Any residual As from the media would have been removed by washing the cells prior to extraction (5) and by partitioning into the aqueous phase (6) during the 3 phenol:chloroform steps in the extraction. If As was incorporated into a lipid or protein it would have partitioned into the phenol, phenol:chloroform, or chloroform fractions (7). Additionally, DNA extracted in this manner on other samples was also successfully used in further analyses, including PCR (8), that require highly purified DNA (9).
The arsenic measured by NanoSIMS in the gel band is consistent with our other measurements and another line of evidence.
Our radiolabeled 73AsO43- experiment showed that of the total radiolabel associated with the cell pellet 11.0 % ± 0.1 % was associated with the DNA/RNA fraction (10). This indicated that we should expect some arsenate of the total pool associated with the nucleic acids (11). To interpret these data, we coupled our interpretation with our EXAFS evidence suggesting that intracellular arsenic was As(V) bound to C (12), and was not free in solution as an ion. This suggests the As is in, an organic molecule with bond distances consistent with a chemical environment analogous to phosphate (Figs. 3A, S3 "bond lengths" table). Further supporting our interpretation of the previous mentioned two analyses, we used a third line of evidence from NanoSIMS, a completely different technique from the other two. We find elemental arsenic (as measured by NanoSIMS) associated with the gel band that is more than two times the background in the gel (13). Based on the above discussion, we do not feel this is a valid concern.
My concerns:
(1) The DNA extraction procedure included only some of the components of a standard DNA extraction protocol. First, only a single ethanol precipitation was done, whereas getting relatively pure DNA requires at least two rounds of extraction and precipitation. Second, the pellets were not washed, so that unincorporated arsenate (or phosphate) present in the aqueous fractions may have been precipitated with the DNA, and may also have been present in the alcohol supernatants contaminating the pellets. Third, no column clean-up step was done.
(2) This statement implies that unincorporated arsenate in the cell lysate would partition into the phenol and chloroform. This seems a priori improbable, as arsenate is very soluble in water. No controls were done to find out how unincorporated arsenate or phosphate would partition in these extractions.
(3) Gel electrophoresis can remove impurities but it is not guaranteed to do so. Any impurities that migrate at a similar rate to the DNA, or are electrostatically associated with it, will be present in the gel slice. Any impurities that diffuse into the gel buffer may become distributed throughout the gel. Any impurities already present present in the agarose or gel buffer will also be in the gel slices. These concerns are strengthened by the failure to purify the DNA away from the gel slice (see (13) below).
(4) No control was performed for non-covalent association of arsenate (or phosphate) with DNA. In another control extraction, arsenate and lysis solution should have been mixed with previously purified DNA from E. coli or other phosphate-grown cells, to see if any arsenic co-purified with the DNA.
(5) No control was performed for the effectiveness of this washing. E. coli or other cells grown in the absence of arsenate should have been mixed with the +As/-P medium and then subject to the same washing and extraction steps.
(6) Here arsenate is predicted to partition into the aqueous phase. Is this the same arsenate that partitioned into the phenol and chloroform in (2) above?
(7) In the ICP-MS analysis presented in Table S1, almost all the arsenic did partition into the phenol phase, and almost as much arsenic was present in the phenol fraction of the phosphate-grown cell (4725 vs 3683 ppb). In fact, the aqueous phase of arsenate-grown cells contained no detectable arsenic at all, even though this is the fraction from which the arsenic-containing DNA was precipitated.
(8) The online Methods say that DNA from all growth conditions, worked fine in the PCR reactions used for the phylogenetic analysis. This strongly suggests that the DNA from arsenate-grown cells has a normal phosphorus backbone. The polymerases used for PCR have very high fidelity and would not tolerate substitution of arsenic for phosphorus.
(9) PCR does not require purified DNA; it even works very well on whole-cell lysates.
(10) See point (7)
(11) But this pool is expected to contain all of the water-soluble constituents of the cell. The elemental analysis report that the arsenic was bonded to carbon doesn't mean that it is bonded to DNA. And most of this arsenic partitioned into the phenol phase - is it thought to be lipid? If so, maybe the arsenic is bonded to C in lipids.
(12) Wait! IANAC (I am not a chemist), but if the 'intracellular arsenic was As(V) bound to C' then it couldn't be arsenic incorporated into DNA or RNA, as it would then have to be bound to O in DNA's diester backbone.
(13) Let's think more about the arsenic in the gel bands. The whole gel slices were assayed (the DNA was not purified away from the agarose); since the gel is 1% agarose and a gel slice is unlikely to weigh less than 100 mg, each slice would contain at least 1 mg of agarose. The DNA bands in Fig. 2A are unlikely to contain more than 1 µg of DNA, probably less for arsenate-grown DNA in lane 2). Thus we can generously assume that 99.9% of the carbon in each DNA sample came from the agarose, and no more than 0.1% from the DNA. According to the figure legend and the numbers at the bottoms of the gel lanes the arsenate-grown sample had 13.4 atoms of arsenic per 10^6 atoms of carbon. This is 13.4 arsenic atoms per 1000 DNA carbons. Since A, G and T nucleotides contains 10 carbons and C has 9, this is 13.4 arsenic atoms per 102.6 nucleotides, or about 26 per 100 base pairs. That's quite a lot of arsenic. Even more surprising, the phosphate-grown sample had 6.9 arsenics per 10^6 carbons, which would be about 14 arsenics per 100 bp. The gel blank had even more arsenic, and three times as much phosphorus. This strongly suggests that the gel was contaminated with both arsenic and phosphorus, perhaps introduced with the DNA samples. Until such contamination can be ruled out, the two-fold higher arsenic concentration and three-fold lower phosphorus concentration associated with the arsenic-grown DNA sample cannot be seen as significant.
Question:
Others have argued that arsenate-linked DNA should have quickly fallen apart when exposed to water. Could you address this?
Answer:
We are not aware of any studies that address arsenate bound in long chain polyesters or nucleotide di- or tri-esters of arsenate, which would be directly relevant to our study. Published studies have shown that simple arsenic esters have much higher hydrolysis rates than phosphate esters (1-3). The experiments published to date have specifically looked at the exchange or hydrolysis of alkyl tri-esters of arsenate [Eqn. 1] and alkyl di-esters of arsenite [Eqn. 2]:
OAs(OR)3 + H2O → OAs(OH)(OR)2+ ROH [1]
OAs(OH)(OR)2 + H2O → OAs(OH)2(OR) + ROH [2]
where R = methyl, ethyl, n-pentyl and isopropyl. Reference 2 demonstrated that the hydrolysis rates for these simple alkyl triesters of arsenate decreased with increasing carbon chain length (complexity) of the alkyl substituent (methyl > ethyl > n-pentyl > isopropyl) (14). No work has been done on the hydrolysis rates of arsenate-linked nucleotides or other biologically relevant moieties.
If the hydrolytic rate trend reported in Ref. 2 continues to larger-weight organics, such as those found in biomolecules, it is conceivable that arsenate-linked biopolymers might be more resistant to hydrolysis than previously thought (15). The small model compounds investigated in Refs. 1-3 are relatively flexible and can easily adopt the ideal geometry for water to attack the arseno-ester bond (16). Arsenate esters of large, bio-molecules, however, are likely to be more sterically hindered leading to slower rates of hydrolysis (17).
This type of steric constraint on reaction rate accounts for the wide range of rates seen in the behavior of some phosphate linked nucleotides. In small ribozymes, the phophodiester linkages at the site of catalysis can be hydrolyzed on the order of tens of seconds (with a chemical rate of 1 s-1). This rate enhancement is achieved by orienting the linkage for in-line attack by a nucleophile (an adjacent 2' hydroxyl group). Moreover, the autodegradation patterns are consistent with specific base composition. On the other hand, the hydrolysis rates for phosphodiester bonds in A form duplexes of RNA are many orders of magnitude slower, because these linkages cannot access easily the geometry necessary for hydrolysis.
The rates in DNA may be much slower than model compounds because of the geometrical constraints imposed upon the backbone by the helix (18).
The kinetics of the hydrolysis of arsenate-linked biopolymers is clearly an area where more research is warranted.
My concerns:
(14) Again IANAC. But note that these are hydrocarbons and thus quite hydrophobic, especially the pentyl chain (5 carbons) and the isopropyl chain (3 branching carbons). Attaching three of these by ester bonds to the arsenate effectively surrounds the bonds with hydrophobic shells that exclude water. Thus it's not surprising that the hydrolysis reaction occurs less often.
(15) But if the increased stability described in Reference 2 (yes, I looked at this paper, and I'm trying to get hold of the 1870 reference too) is due to increasing hydrophobicity of the ester bonds' environment, then the effect will not extrapolate to long hydrophilic biological molecules such as DNA.
(16) Not if they're surrounded by a hydrophobic shell, and probably also associating with each other to reduce the shell's exposure to the aqueous solvent.
(17) DNA has its backbone on the outside of the double helix, and the entire molecule is quite hydrophilic.
(18) Assuming the overall structure of arsenic bonds in DNA is like that of phosphorus bonds in DNA, shouldn't any stability-enhancing geometrical constraints of the DNA structure be experienced by both phosphorus and arsenic bonds? We would then still expect arsenic bonds in DNA to be 100-fold less stable than phosphate bonds.
Question:
Is it possible that salts in your growth media could have provided enough trace phosphorus to sustain the bacteria?
Answer:
The data and sample labeling in Table S1 has caused some confusion. To clarify, for every experiment, a single batch of artificial Mono Lake water was made with the following formulation: AML60 salts, no P, no As, no glucose, no vitamins. Table S1 shows examples of ICPMS measurements of elemental phosphorus (~3 µM) and arsenate made on this formulation prior to any further additions (19, 20). Then we added glucose and vitamins for all three treatments and either As for the +As treatments or P for the +P treatments. The P measurements made on the medium after the addition of sucrose and vitamins and after addition of As were also ~3 µM in this batch. Therefore, it was clear that any P impurity that was measured (~3 µM, this was the high range) came in with the major salts, and that all experiments contain identical P background (including any P brought in with the culture inocula).
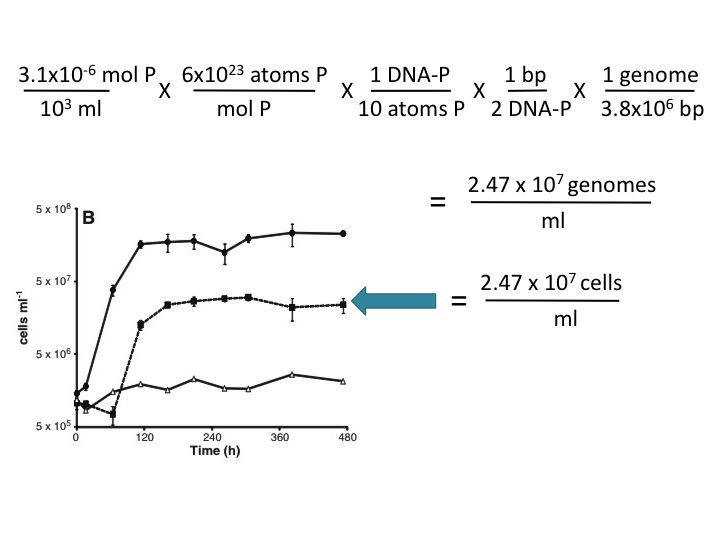
In the Science paper, we show data from one experiment of many replicated experiments that demonstrates no growth of cells in media without added arsenate or phosphate (Figure 1). These data clearly demonstrate that strain GFAJ-1 was unable to utilize the 3µM P to support further growth in the absence of arsenate (21). Moreover, the intracellular P content determined for the +As/-P grown cells was not enough to support the full requirement of P for cellular function (22).
Note on culturing: All experiments were initiated with inocula from sustained +As/-P conditions. Prior to the experiments, the cells had been grown long term, for multiple generations from a single colony grown on solid media with no added phosphate. Before this, they were grown as an enrichment for more than 10 transfers and always into new medium that was +As/-P. We therefore feel that there is not significant carry-over of P. We also argue that there would not have been enough cellular P to support additional growth based on an internal recycling pool of P (23).
My concerns:
(19) The two batches of AML60 salts assayed contained 3.7 and <0.3 µM P, and the single batch of cell wash solution contained 7.4 µM P. Given this variability, the similarity of the two batches of -P/+As medium doesn't inspire much confidence.
(20) Was no effort made to identify and eliminate the source(s) of this contamination?
(21) Agreed, with the proviso that the media be tested and shown to be identical except for the phosphate and arsenate. But this wouldn't mean that arsenic replaced phosphorus in any biological molecules in GFAJ cells, just that the cells needed arsenate for something.
(22) Did this calculation take into account the very high carbon content of the poly-hydroxybutyrate granules in these cells? PHB can account for up to 90% of the dry weight of phosphate-starved cells, and its carbon will skew estimates of C:P ratios.
(23) This assertion is not supported here by any evidence, and it is contradicted by the Makino et al. (2003) reference cited by the Science paper (ref. 13). These authors found that 10% of the P in phosphate-limited E. coli is in the DNA and the rest is in RNA and other cellular components. Using this value and your own estimate of genome size (3.8 Mbp), ~3 µM P is sufficient to account for the observed growth in the -P/+As medium. Here's the calculation: