














I've run a couple of passes through the entire genome. The results are just as unsurprising as I had expected (is that a
 tautology?). Blogger won't upload the logo image files right now so I'll add them later (done). Basically, the forward and reverse motifs are fairly close reverse complements of each other. I was going to write that this means that there are no strong effects of the direction of DNA replication, but that's not true. To see effects of the direction of
tautology?). Blogger won't upload the logo image files right now so I'll add them later (done). Basically, the forward and reverse motifs are fairly close reverse complements of each other. I was going to write that this means that there are no strong effects of the direction of DNA replication, but that's not true. To see effects of the direction of 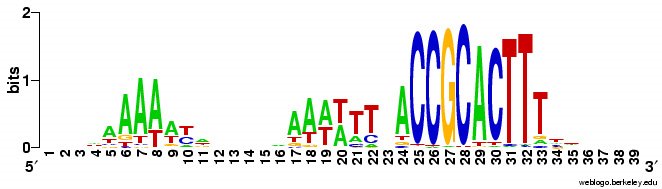 DNA replication I'd need to split the genome sequence into two parts, one clockwise from the origin of replication to the probable terminus, and one counterclockwise from the origin to the terminus and compare the forward and reverse motifs in each half. That way the forward/clockwise and referse/counterclockwise motifs would be derived from the 'leading' strand and the reverse/clockwise and forward/counterclockwise motifs would be derived from the lagging strand. [Or vice versa, as I don't know which strand is which.] Hmm, I don't think anyone has ever done this. So I should.
DNA replication I'd need to split the genome sequence into two parts, one clockwise from the origin of replication to the probable terminus, and one counterclockwise from the origin to the terminus and compare the forward and reverse motifs in each half. That way the forward/clockwise and referse/counterclockwise motifs would be derived from the 'leading' strand and the reverse/clockwise and forward/counterclockwise motifs would be derived from the lagging strand. [Or vice versa, as I don't know which strand is which.] Hmm, I don't think anyone has ever done this. So I should.
I think it would be interesting to see if there is an orientation bias...although I am thinking that we would not predict this to be the case, or that if was the case, it would have nothing to do with DNA uptake.
ReplyDeleteDoesn't the fragmentation mask add a bias to your "un-biased" approach? I guess it as good as it will get.
Re: Heather's comment above, I think that the paper by Smith, Gwinn & Salzberg "DNA uptake signal sequences in naturally transformable bacteria" looks at orientation.
ReplyDeleteI took a quick look at the Smith et al. paper while writing this post. They found that the frequencies of forward- and reverse-orientation USS cores did not differ between the directions of replication. But they didn't look for differences in the flankng sequences or for non-perfect cores. The motif analysis makes it easy to now do that.
ReplyDeleteIt's true that including a fragmentation mask adds a bias. It's not possible to do a truly unbiased search (I think this may be a philosophical issue), and I think the fragmentation mask is the best compromise, as it makes no assumptions about sequence, and the analysis provides a lot of information even about the 'non-significant' positions.