














I continue to entertain the (so far unsupported) idea that USSs are sequences at which DNA can be easily kinked to permit its passage across the bacterial outer membrane. (The animation on our lab home page shows how I imagine this happening.) I've been concerned that thinking only about a USS consensus that's the average of all the real USS might mask some important properties of individual USSs, so I'm also trying to understand the properties of single real USS sequences.
I've just found a web site called MDDNA, which analyzes the predicted structure of any
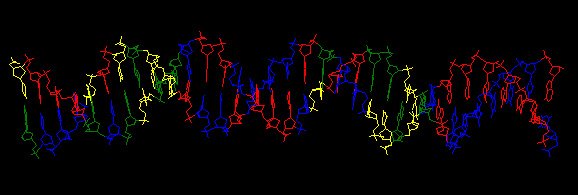 short DNA sequence you paste into it, using the latest predictions of how different combinations of neighbouring bases modulate the standard "B" structure of the DNA double helix. The image at the left shows the structure of a typical USS. This is just a screenshot - the MDDNA analysis creates a "PDB" structure file and lets you view and play with it in 3-D in a "Jmol" applet.
short DNA sequence you paste into it, using the latest predictions of how different combinations of neighbouring bases modulate the standard "B" structure of the DNA double helix. The image at the left shows the structure of a typical USS. This is just a screenshot - the MDDNA analysis creates a "PDB" structure file and lets you view and play with it in 3-D in a "Jmol" applet.The site also provides all the 'helicoidal parameters' associated with the structure it has produced. But I'll need to do some work before I can understand what these mean.
You've probably noticed that the DNA in the image is not bent at all (certainly not sharply kinked as I suggested above). Neither were any of the molecules predicted from the other USS sequences I tried. This might mean that USS sequences are not prone to bending at all (i.e. my idea is wrong). Or it might be that the sequences are straight when free in solution, but bend easily when bound to their receptor (i.e. my idea could be right). I need to keep looking and reading to find out more about the dynamic properties of DNA molecules under stress.
Cool visualization! How much is known about DNA bending?
ReplyDelete